
Síndrome de Fahr secundario a hipoparatiroidismo: una causa infrecuente de movimientos anormales en niños
Resumen
Presentamos un caso de síndrome de Fahr en un escolar de 10 años masculino con movimientos distónicos cervicales y de miembros superiores en quien se sospechó que eran de origen convulsivo. En las imágenes cerebrales se evidenciaron hiperdensidades gangliobasales y subcorticales bilaterales, y las pruebas bioquímicas mostraron hipocalcemia e hiperfosfatemia con paratohormona baja. Recibió tratamiento anticonvulsivante, carbonato de calcio y calcitriol, con mejoría de los síntomas y sin recurrencia de movimientos anormales.
Un caso familiar de microduplicación distal 22q11.2*
Resumen
Introducción: La microduplicación distal 22q11.2 es una entidad rara, pero de la que están apareciendo cada vez más casos en la literatura, ampliando en cada referencia el espectro de manifestaciones. Presentamos dos casos dentro de la misma familia.
Presentación del caso: El caso índice es un recién nacido prematuro, con historia clínica neonatal de sepsis precoz, displasia broncopulmonar, ductus arterioso persistente, hiperbilirrubinemia que precisa exanguinotransfusión, raquitismo grave e hipoacusia neurosensorial profunda bilateral. En su seguimiento evolutivo destacan la talla baja, así como unos rasgos dismórficos, entre los que resaltan macrocefalia con frente amplia, epicanto y braquidactilia. En la RMN de cráneo se detecta ventriculomegalia, sin otras alteraciones, y a los 4 años de edad presenta moderado retraso del lenguaje. Su madre tiene unos rasgos faciales similares, con baja estatura e hiperlordosis, pero sin alteraciones en el aprendizaje. Se realiza hibridación genómica comparativa (aCGH), demostrando una secuencia duplicada de 1,5 Mb en la región 22q11.2, tanto en el paciente como en su madre.
Discusión: La microduplicación distal de la región 22q11.2 se presenta con una amplia variabilidad clínica, tanto interindividual como dentro de una misma familia. Es difícil una sospecha clínica previa, realizándose el diagnóstico gracias al estudio con aCGH
Disgenesia gonadal completa 46 XY: forma atípica con talla baja
Resumen
La disgenesia gonadal completa 46 XY (46, XY CGD) es un trastorno del desarrollo sexual. Se caracteriza por el cariotipo 46 XY, genitales externos femeninos normales, presencia de estructuras müllerianas y gónadas sin desarrollar. Es un síndrome infrecuente, cuyos pacientes tienen un fenotipo femenino normal y una talla normal o alta, por lo que se diagnostican por retraso puberal o amenorrea primaria. La mayoría de los pacientes con 46, XY CGD muestran un gen SRY normal. Asociado a la presencia de un cromosoma Y, existe un riesgo marcado de tumores gonadales, especialmente después de la pubertad. El gonadoblastoma es el tumor más frecuente y tiene un alto riesgo de malignización hacia disgerminoma.
Presentamos el caso de una niña que consulta a los 8 años de edad por talla baja. A la exploración la paciente presenta un fenotipo femenino normal, genitales externos femeninos, con estadio de Tanner I, peso de 21,6 kg (DE -1,43) y talla de 115,4 cm (DE -3,1). El laboratorio reveló test de estimulación con gonadotropina coriónica humana sin respuesta de testosterona y hormona antimülleriana <1 pmol/L. El cariotipo en sangre periférica es informado como 46 XY, con presencia del gen SRY. La resonancia magnética abdominal mostró la presencia de vagina, útero hipoplásico y ausencia de gónadas. Se realiza gonadectomía bilateral laparoscópica. El análisis anatomopatológico confirmó la presencia de gonadoblastoma puro bilateral de ovarios. Los hallazgos permiten confirmar el diagnóstico de 46, XY CGD. La novedad del caso radica en su baja frecuencia de aparición, la edad del diagnóstico y la presentación con una talla baja.
Diagnóstico clínico de sospecha para una nueva mutación en el gen COL5A1 contenido en el panel del síndrome de Marfan-Like
Introducción: El síndrome de Marfan-Like es una patología congénita que no cumple todos los criterios del síndrome de Marfan. La descripción de su fenotipo nos permite realizar un diagnóstico de sospecha.
Caso clínico: Los autores describen las manifestaciones clínicas observadas en una niña que presenta un cambio en el gen COL5A1 no descrito en el momento actual. Estas alteraciones son hipotonía muscular e hiperlaxitud articular generalizada, déficit de atención, escoliosis, pies planos valgos, facies alargada y miopática, inclinación antimongoloide de las comisuras parpebrales, pabellones auriculares despegados, leve exoftalmos y ptosis bilateral, astigmatismo, fisura palatina, micrognatia, paladar ojival, apiñamiento dental, erupción precoz y secuencia alterada en ambas denticiones.
Conclusión: La sospecha diagnóstica de estas anomalías se basa en los hallazgos clínicos, de ahí la importancia del conocimiento de los mismos para implementar cuanto antes un tratamiento efectivo en la esfera estomatológica.
Fusobacterium necrophorum: una etiología poco frecuente, pero en aumento, de la mastoiditis aguda en niños pequeños
Introducción: Aunque la mastoiditis aguda está causada principalmente por Streptococcus pneumoniae, Staphylococcus aureus y Streptococcus pyogenes, en los últimos años se aprecia un aumento en la incidencia de Fusobacterium necrophorum, especialmente en lactantes.
Caso clínico 1: Lactante que acude a urgencias por fiebre, decaimiento y rechazo de la alimentación. Está taquicárdico, con afectación del estado general, leve palidez cutánea y rigidez de nuca, siendo el resto de la exploración física normal. Con la sospecha de sepsis/bacteriemia se inicia cefotaxima i.v. Pasadas 2 horas aparece otorrea izquierda, y pasadas 36 horas mastoidismo. La tomografía computarizada (TC) craneal pone de manifiesto una otomastoiditis aguda bilateral con absceso subperióstico izquierdo, por lo que el paciente precisa la realización de una mastoidectomía izquierda y un drenaje del absceso subperióstico. En el cultivo del absceso subperióstico se aísla F. necrophorum.
Caso clínico 2: Lactante que acude a urgencias por persistencia de la fiebre y aparición de otorrea a las 24 horas de estar recibiendo amoxicilina-clavulánico v.o. debido a una otitis media aguda derecha. Presenta buen estado general, las constantes son normales y en la exploración física destaca otorrea izquierda con mastoidismo. La TC craneal confirma una otomastoiditis bilateral con posible absceso subperióstico izquierdo. Se realiza una mastoidectomía izquierda y un drenaje del absceso subperióstico. En el cultivo del absceso subperióstico se aísla F. necrophorum.
Conclusión: Ante un lactante con mastoiditis aguda debemos tener en mente el F. necrophorum como posible agente etiológico, asociado con una peor evolución, una mayor necesidad de tratamiento quirúrgico y un mayor riesgo de complicaciones que otros gérmenes.
Caso clínico de enfermedad de Gaucher tipo I y sacroileítis bilateral
La enfermedad de Gaucher (EG) es una enfermedad metabólica rara y hereditaria de almacenamiento lisosómico, cuyo patrón hereditario es autosómico recesivo y está causada por mutaciones en el gen GBA1. En este artículo se describe retrospectivamente el caso clínico de una mujer diagnosticada a los 2,5 años de EG tipo I en el Hospital Universitario de Jerez, cuya evolución se ha seguido en el mismo centro durante más de 20 años. Tras haber estado durante 5 años –aunque de manera interrumpida, debido al desabastecimiento de la enzima– con terapia enzimática sustitutiva (imiglucerasa), desde 2011 (participación en el estudio ENCORE) está siendo tratada con eliglustat tartrato, una terapia de reducción de sustrato. La respuesta al tratamiento es buena y alcanza los objetivos terapéuticos. La sacroileítis bilateral persiste, aunque permanece estable y controlada.
Calcificaciones cerebrales en el diagnóstico precoz del síndrome de DiGeorge sin cardiopatía
Forma de presentación «prono-doloroso-like» del síndrome de Parsonage-Turner
Resultados de aplicar durante 13 años el protocolo de cribado universal de la hipoacusia en recién nacidos y estudio de los casos que no superan el cribado
Inesperada reagudización de enfermedad de Kawasaki
Os supranavicular en el paciente pediátrico
El os supranavicular es un hueso accesorio del pie, poco frecuente, situado en la cara superior de la articulación talonavicular. Raramente sintomático, provoca dolor en el dorso del pie y se diagnostica a menudo de forma errónea como una fractura por avulsión, por lo que es importante tener un conocimiento anatómico adecuado y realizar un buen diagnóstico diferencial. Presentamos el caso de un niño de 8 años de edad, con hueso supranavicular sintomático bilateral, que acude a nuestra consulta por un dolor mecánico en la región dorsal de ambos pies, que se intensifica tras la actividad física. Es fundamental realizar una anamnesis y una exploración física completas para establecer una adecuada orientación diagnóstica. El diagnóstico final se logra mediante la realización de pruebas complementarias, principalmente la proyección lateral de una radiografía simple de ambos pies en carga. El objetivo del tratamiento conservador (ortesis plantar, fundamentalmente) es establecer la función óptima del pie durante las actividades de carga. Si éste no proporciona un alivio tras 4-6 meses de seguimiento, debería considerarse el tratamiento quirúrgico, que consiste en la escisión simple del hueso accesorio.
Enfermedad de Vogt-Koyanagi-Harada: una causa rara de uveítis
La enfermedad de Vogt-Koyanagi-Harada es una uveítis granulomatosa difusa bilateral, asociada a poliosis, vitíligo, alopecia y síntomas neurológicos y auditivos. La etiología es probablemente autoinmune contra los melanocitos, determinada por factores genéticos. Es una entidad muy rara en la edad pediátrica. Presentamos un nuevo caso en un paciente de 12 años de edad.
Respuesta inmunológica de niños celiacos a la vacuna frente a la hepatitis B
Fractura lumbar como presentación de un linfoma linfoblástico de células B
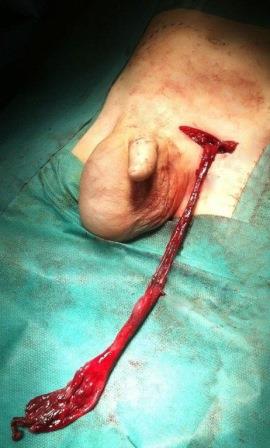
Un caso particular de hidrocele abdominoescrotal bilateral
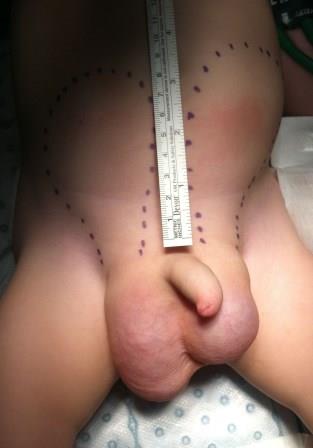 Figura 1. Se palpan dos tumoraciones de consistencia quística a ambos lados de la línea media, que se continúan por la región inguinal hacia el escroto; el paciente presenta genitales externos con hidroceles bilaterales a tensión
Figura 1. Se palpan dos tumoraciones de consistencia quística a ambos lados de la línea media, que se continúan por la región inguinal hacia el escroto; el paciente presenta genitales externos con hidroceles bilaterales a tensión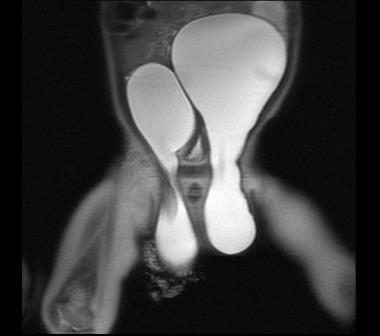
Agenesia del conducto deferente en un paciente con criptoquidia y fibrosis quística. Exposición de un caso y revisión de la bibliografía
Presentamos el caso de un varón de 20 meses de edad con fibrosis quística (ΔF508/ΔF508) y agenesia del conducto deferente como hallazgo durante una orquidopexia. Se realiza una revisión bibliográfica de las alteraciones genéticas observadas en los pacientes con ausencia congénita bilateral y unilateral del conducto deferente (ACBCD y ACUCD), así como el papel del gen regulador de la conductancia transmembrana de la fibrosis quística en otros aspectos relacionados con la fertilidad.
Piomiositis con sacroileítis asociada
La piomiositis es la infección por bacterias del músculo esquelético, infrecuente pero con una incidencia en aumento, por lo que se debe tener en cuenta a la hora de realizar el diagnóstico diferencial de la impotencia funcional febril en los miembros inferiores en la edad pediátrica. Se presenta el caso de una niña diagnosticada de piomiositis con sacroileítis asociada tratada en nuestro hospital, y se ha realizado una revisión bibliográfica a propósito del tema.
Enfermedad multiquística pulmonar en el niño. Diagnóstico diferencial
Presentamos el caso de un varón de 14 años, diagnosticado de histiocitosis de células de Langerhans a los 16 meses de edad, en el que, sin presentar patología respiratoria evidente, se aprecia un patrón multiquístico bilateral en la tomografía computarizada de tórax, y un patrón intersticial bilateral en la radiografía convencional. Atendiendo a las características radiológicas del paciente, proponemos el diagnóstico diferencial de entidades infrecuentes que cursan con expresión multiquística pulmonar en la edad pediátrica.
Treclinac®: Nueva y única combinación para el tratamiento del acné

Una nueva formulación que combina clindamicina y tretinoína consigue rápidos resultados contra el acné
Treclinac® es un nuevo producto para el acné que trata eficazmente tanto las lesiones inflamatorias como las no inflamatorias y que posee muy buen perfil de tolerabilidad. Estas propiedades son debidas a una formulación innovadora que contiene clindamicina y dos formas de tretinoína: una forma solubilizada, disponible inmediatamente y una forma cristalina en suspensión, que permite una lenta y progresiva penetración del retinoide en la piel1.
Treclinac® posee importantes beneficios frente a las combinaciones actualmente disponibles para el tratamiento del acné:
Treclinac® es más eficaz que la combinación clindamicina / peróxido de benzoilo puesto que, al poseer esta última dos agentes antimicrobianos y no contar con un retinoide en su formulación, no actúa en las lesiones no inflamatorias2-4.
Treclinac® es mejor tolerado que otras combinaciones a base de retinoide, como la que contiene adapaleno y peróxido de benzoilo, manteniendo la misma eficacia frente a lesiones inflamatorias y no inflamatorias5,6.
Treclinac® es fácil de utilizar y no produce blanqueamiento del cabello o los tejidos ya que no contiene peróxido de benzoilo como las otras dos combinaciones existentes.
Treclinac® cumple con las recomendaciones del Grupo Alianza Global para Mejorar los Resultados en el Acné, que establece que la mayoría de los pacientes con acné deberían ser inicialmente tratados con un retinoide tópico en combinación con un agente antimicrobiano puesto que esto permite atacar a la mayor parte de los factores patogénicos del acné y trata tanto las lesiones inflamatorias como las no inflamatorias7.
Más información:
info@meda.es
www.meda.es
Acceso a ficha técnica
Bibliografía
- Del Rosso JQ, Jitpraphai W, Bhambri S, et al. Clindamycin phosphate 1.2%- tretinoin 0.025% gel: vehicle characteristics, stability, and tolerability. Cutis 2008;81:405-8.
- Schlessinger J, Menter A, Gold M, et al. Clinical safety and efficacy studies of a novel formulation combining 1.2% clindamycin phosphate and 0.025% tretinoin for the treatment of acne vulgaris. J Drugs Dermatol 2007;6:607-15.
- Bettoli V. Efficacy and safety of novel clindamycin 1% / tretinoin 0.025% formulation for acne vulgaris: pooled analysis of 3 phase III studies. Presented at AAD 2013; Poster 6404.
- Duac. Summary of product characteristics. 2011; Stiefel (a GlaxoSmithKline Company), UK.
- Goreshi R, Samrao A, Ehst BD. A double-blind, randomized, bilateral comparison of skin irritancy following application of the combination acne products clindamycin/tretinoin and benzoyl peroxide/adapalene. J Drugs Dermatol 2012;11:1422-6.
- Perez M. Cross-study comparison of efficacy and safety of clindamycin 1%/tretinoin 0.025% and adapalene 0.1%/benzoyl peroxide 2.5% for acne vulgaris. Presented at AAD 2013; Poster 6087.
- Thiboutot D, Gollnick H, Bettoli V, et al. New insights into the management of acne: an update from the Global Alliance to Improve Outcomes in Acne group. J Am Acad Dermatol 2009;60:S1-50.
Asma de control difícil como manifestación de una traqueomalacia
Sr. Director:
El asma de control difícil (ACD) se define como aquella insuficientemente controlada pese a un tratamiento apropiado, ajustado a la gravedad clínica. De los pacientes considerados como ACD, sólo una pequeña proporción lo son en realidad1,2.
Según la guía GEMA, el diagnóstico final de ACD exige tres condicionantes previos: a) verificar que el tratamiento es el adecuado y se cumple correctamente; b) descartar otras enfermedades que se asemejan al asma, y c) asegurar el control de los factores agravantes de la enfermedad (ácaros del polvo, humo del tabaco, estrés)1. Algunas de las enfermedades que pueden manifestarse con síntomas respiratorios que simulen asma son las siguientes: laringo-tráqueo-broncomalacia, laringotraqueobronquitis, disfunción de las cuerdas vocales, fístula traqueoesofágica, cuerpo extraño bronquial, tumor de las vías aéreas inferiores, compresión extrínseca de la vía aérea, fibrosis quística, enfermedad respiratoria por reflujo gastroesofágico (RGE) o discinesia ciliar primaria2-4. Para confirmar el diagnóstico de asma es útil la realización de una espirometría forzada cuando se observa un patrón obstructivo con valores de FEV1 y FEV1/FVC bajos e incremento del primero >12% tras el uso de un broncodilatador1.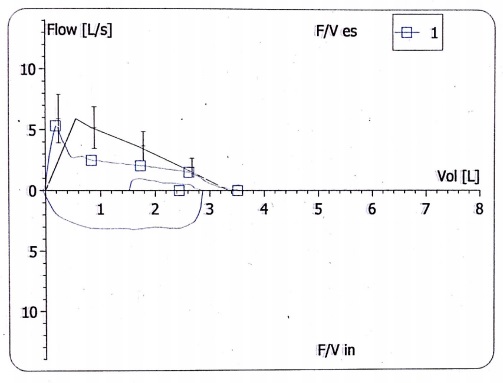 Figura 1. Curva flujo-volumen. Pico de flujo espiratorio precoz con un brusco descenso inicial y una pendiente aplanada hasta el final de la maniobra. Valores: FVC 3 L (100% del teórico), FEV1 2,3 L (93,4%), FEV1/FVC 77,8% (92%), PEF 5,2 L/s (96%), MMEF 75/25 2 L/s (68%), TLC-He 3,9 L (98%), RV-He 0,9 L (101%), RV/TLC-He 23% (96%). Sin cambios tras la administración de salbutamolPresentamos el caso de un niño de 12 años de edad, remitido a nuestro servicio para su valoración, diagnosticado de asma no alérgica desde que tenía 1 año, y de difícil control por persistir ésta de forma sintomática a pesar del tratamiento. En el periodo neonatal presentó estridor con la tos y el llanto, que fue atribuido a una laringomalacia. Sólo refiere un ingreso por crisis asmática, y ha presentado dos crisis de broncoespasmo anuales controladas ambulatoriamente. En los periodos intercrisis constataba una tos nocturna que no interfería con el sueño, y tos con el ejercicio físico, sin limitación del mismo. No refiere rinoconjuntivitis, otitis, vómitos, dolor abdominal o alteración del apetito, ronquido nocturno o apneas. Tampoco presenta tabaquismo activo ni pasivo. El tratamiento que toma es montelukast 5 mg y salmeterol/fluticasona 25/50 µg inhalada sin cámara (2 pulsaciones cada 12 h). Tanto el paciente como el padre niegan olvidos u omisiones del tratamiento. En la exploración clínica se observan unos percentiles de peso y talla de 95 y 25, respectivamente. Presenta buen color e hidratación. No se observa tiraje ni acropaquias. En la auscultación se observa una buena entrada de aire bilateral y un estridor inspiratorio con maniobras profundas, audible predominantemente en el hemitórax izquierdo. Las pruebas complementarias realizadas inicialmente fueron una radiografía de tórax, con resultado normal, y una espirometría forzada. La curva flujo-volumen (CFV) (figura 1) pone de manfiesto una disminución rápida del flujo espiratorio forzado tras el pico inicial en relación con el colapso de la vía aérea central, seguido de un flujo espiratorio disminuido en meseta, compatible con una traqueomalacia5. La prueba broncodilatadora fue negativa. Se programó una fibrolaringobroncoscopia (figura 2) que confirmó el diagnóstico de sospecha (traqueomalacia) y, además, la presencia de un edema de aritenoides. Este último hallazgo sugería la presencia de RGE, por lo que se añadió al tratamiento omeprazol 20 mg/día. A las 6 semanas, en ausencia de cambios clínicos y funcionales, se suspendió el omeprazol y se desescaló la medicación antiasmática a propionato de fluticasona 100 µg/12 h con cámara como paso previo a la suspensión definitiva. Seis meses después no ha presentado disnea ni infecciones de la vía aérea superior, tolera bien el ejercicio y presenta una auscultación pulmonar normal. En su última visita (14 años) no tomaba tratamiento médico y no refería deterioro clínico. En la espirometría de control no se apreciaron cambios respecto a las previas.
Figura 1. Curva flujo-volumen. Pico de flujo espiratorio precoz con un brusco descenso inicial y una pendiente aplanada hasta el final de la maniobra. Valores: FVC 3 L (100% del teórico), FEV1 2,3 L (93,4%), FEV1/FVC 77,8% (92%), PEF 5,2 L/s (96%), MMEF 75/25 2 L/s (68%), TLC-He 3,9 L (98%), RV-He 0,9 L (101%), RV/TLC-He 23% (96%). Sin cambios tras la administración de salbutamolPresentamos el caso de un niño de 12 años de edad, remitido a nuestro servicio para su valoración, diagnosticado de asma no alérgica desde que tenía 1 año, y de difícil control por persistir ésta de forma sintomática a pesar del tratamiento. En el periodo neonatal presentó estridor con la tos y el llanto, que fue atribuido a una laringomalacia. Sólo refiere un ingreso por crisis asmática, y ha presentado dos crisis de broncoespasmo anuales controladas ambulatoriamente. En los periodos intercrisis constataba una tos nocturna que no interfería con el sueño, y tos con el ejercicio físico, sin limitación del mismo. No refiere rinoconjuntivitis, otitis, vómitos, dolor abdominal o alteración del apetito, ronquido nocturno o apneas. Tampoco presenta tabaquismo activo ni pasivo. El tratamiento que toma es montelukast 5 mg y salmeterol/fluticasona 25/50 µg inhalada sin cámara (2 pulsaciones cada 12 h). Tanto el paciente como el padre niegan olvidos u omisiones del tratamiento. En la exploración clínica se observan unos percentiles de peso y talla de 95 y 25, respectivamente. Presenta buen color e hidratación. No se observa tiraje ni acropaquias. En la auscultación se observa una buena entrada de aire bilateral y un estridor inspiratorio con maniobras profundas, audible predominantemente en el hemitórax izquierdo. Las pruebas complementarias realizadas inicialmente fueron una radiografía de tórax, con resultado normal, y una espirometría forzada. La curva flujo-volumen (CFV) (figura 1) pone de manfiesto una disminución rápida del flujo espiratorio forzado tras el pico inicial en relación con el colapso de la vía aérea central, seguido de un flujo espiratorio disminuido en meseta, compatible con una traqueomalacia5. La prueba broncodilatadora fue negativa. Se programó una fibrolaringobroncoscopia (figura 2) que confirmó el diagnóstico de sospecha (traqueomalacia) y, además, la presencia de un edema de aritenoides. Este último hallazgo sugería la presencia de RGE, por lo que se añadió al tratamiento omeprazol 20 mg/día. A las 6 semanas, en ausencia de cambios clínicos y funcionales, se suspendió el omeprazol y se desescaló la medicación antiasmática a propionato de fluticasona 100 µg/12 h con cámara como paso previo a la suspensión definitiva. Seis meses después no ha presentado disnea ni infecciones de la vía aérea superior, tolera bien el ejercicio y presenta una auscultación pulmonar normal. En su última visita (14 años) no tomaba tratamiento médico y no refería deterioro clínico. En la espirometría de control no se apreciaron cambios respecto a las previas.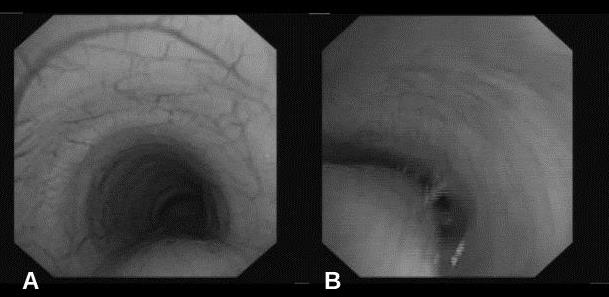 Figura 2. Fibrobroncoscopia dinámica. Visión de la tráquea. A) En la fase inspiratoria se aprecia una luz uniforme en toda su longitud. B) En la fase espiratoria se aprecia un colapso de la luz traqueal por protrusión anterior de la pars membranosaLa traquebroncomalacia se caracteriza por una debilidad de la pared y una disminución dinámica de la luz de la tráquea y/o bronquios, especialmente durante la espiración6. La malacia puede afectar a toda la tráquea o estar localizada, en cuyo caso hay que considerar la posibilidad de una compresión extrínseca o una secuela por anomalía congénita (atresia de esófago). La dificultad para la salida de aire y las secreciones durante la espiración se plasman clínicamente en estertores, sibilancias, estridor, intolerancia al ejercicio, tos, infecciones recurrentes de las vías respiratorias inferiores y atrapamiento aéreo. El grado de malacia determina la severidad de los síntomas, y en los casos más graves puede producir apnea y cianosis. Los grados leve o moderado se acompañan de síntomas similares a los presentes en el asma, por lo que cuando se trata erróneamente como tal, la respuesta a los broncodilatadores y corticoides es escasa o nula, confundiéndose con un ACD. Como consecuencia de ello, se pautan tratamientos antiasmáticos prolongados y se demora el tratamiento de las infecciones de las vías aéreas inferiores7.
Figura 2. Fibrobroncoscopia dinámica. Visión de la tráquea. A) En la fase inspiratoria se aprecia una luz uniforme en toda su longitud. B) En la fase espiratoria se aprecia un colapso de la luz traqueal por protrusión anterior de la pars membranosaLa traquebroncomalacia se caracteriza por una debilidad de la pared y una disminución dinámica de la luz de la tráquea y/o bronquios, especialmente durante la espiración6. La malacia puede afectar a toda la tráquea o estar localizada, en cuyo caso hay que considerar la posibilidad de una compresión extrínseca o una secuela por anomalía congénita (atresia de esófago). La dificultad para la salida de aire y las secreciones durante la espiración se plasman clínicamente en estertores, sibilancias, estridor, intolerancia al ejercicio, tos, infecciones recurrentes de las vías respiratorias inferiores y atrapamiento aéreo. El grado de malacia determina la severidad de los síntomas, y en los casos más graves puede producir apnea y cianosis. Los grados leve o moderado se acompañan de síntomas similares a los presentes en el asma, por lo que cuando se trata erróneamente como tal, la respuesta a los broncodilatadores y corticoides es escasa o nula, confundiéndose con un ACD. Como consecuencia de ello, se pautan tratamientos antiasmáticos prolongados y se demora el tratamiento de las infecciones de las vías aéreas inferiores7.
El caso presentado corresponde probablemente a una traqueomalacia congénita leve, tratada durante años como asma y, finalmente, considerada como ACD antes de remitir al paciente a nuestro servicio.
La malacia de las vías aéreas se asocia frecuentemente a RGE8. La relajación transitoria del esfínter esofágico inferior respondería a un mecanismo reflejo desencadenado por la distensión del esófago, secundaria al paso de aire durante la espiración, y del estómago, por el aire previamente deglutido9. La presencia de edema de aritenoides en el caso presentado sugiere un RGE, aunque el tratamiento antiácido no se acompañó de una mejoría clínica constatable.
El diagnóstico definitivo de traqueomalacia se basa en la visualización del colapso dinámico de la vía aérea. En los adultos se puede realizar una tomografía computarizada en inspiración y espiración forzada, aunque la fibrobroncoscopia constituye la técnica de elección en los niños, cuya colaboración es limitada5,6. En los casos de ACD es necesario reconsiderar el diagnóstico, realizando una espirometría forzada antes de investigar otras causas3. La CFV es sugestiva de traqueomalacia cuando presenta un descenso rápido del pico de flujo inicial, seguido de una meseta prolongada espiratoria10.
Pese a la creencia inicial de resolución clínica y funcional de la traqueobroncomalacia en los primeros años de vida, la mayoría de los niños diagnosticados endoscópicamente presentan síntomas persistentes y una alteración espirométrica (obstructiva) hasta, al menos, el inicio de la adolescencia5. En el caso presentado se observa una mejoría progresiva de la clínica con el tiempo, con o sin tratamiento antiasmático. Puede que esta evolución sea casual o que los síntomas, aunque presentes, no sean interpretados como patológicos por parte del paciente y, por tanto, no los comunique.
En conclusión, la CFV de la espirometría forzada en los pacientes evaluados por ACD puede orientar al diagnóstico de traqueomalacia, aunque es necesaria la confirmación endoscópica.
Bibliografía
- Plaza V. coordinador. Gema 2009. Guía española para el manejo del asma. Disponible en: http://www.gemasma.com
- Navarro M, Andrés A, Asensio O, García ML, Liñán S, Villa JR. Guía de diagnóstico y tratamiento del asma de control difícil en el niño. An Pediatr. 2009; 71: 548-567.
- Bush A, Saglani S. Management of severe asthma in children. Lancet. 2010; 376: 814-825.
- Weinberger M, Abu-Hasan M. Pseudo-asthma: when cough, wheezing, and dyspnea are not asthma. Pediatrics. 2007; 120: 855-864.
- Moore P, Smith H, Greer R, McElrea M, Masters I. Pulmonary function and long-term follow-up of children with tracheobronchomalacia. Pediatr Pulmonol. 2012; 47: 700-705.
- Majid A, Fernández L, Fernández-Bussy S, Herth F, Ernst A. Traqueobroncomalacia. Arch Bronconeumol. 2010; 46: 196-202.
- Boogaard R, Huijsmans SH, Pijnenburg MW, Tiddens HA, De Jongste JC, Merkus PJ. Tracheomalacia and bronchomalacia in children: incidence and patient characteristics. Chest. 2005; 128: 3.391-3.397.
- Yalçın E, Doğru D, Özçelik U, Kiper N, Tana Aslan A, Gözaçan A. Tracheomalacia and bronchomalacia in 34 children: clinical and radiologic profiles and associations with other diseases. Clin Pediatr. 2005; 44: 777-781.
- Turbyville JC. Applying principles of physics to the airway to help explain the relationship between asthma and gastroesophageal reflux. Med Hypotheses. 2010; 74: 1.075-1.080.
- Majid A, Sosa AF, Ernst A, Feller-Kopman D, Folch E, Singh A, et al. Pulmonary function and flow volume loop patterns in patients with tracheobronchomalacia. Respir Care. 2013 [consultado el 20 julio de 2013]. Disponible en: http://rc.rcjournal.com/content/early/2013/03/12/respcare.02277.full.pdf+html
II Reunión Nacional de Nefrourología Pediátrica. Puesta al día en el diagnóstico prenatal de anomalías estructurales del riñón y las vías urinarias
Objetivo: Precisar las indicaciones quirúrgicas y la realización de pruebas diagnósticas en pacientes con anomalías estructurales del riñón y las vías urinarias de diagnóstico prenatal.
Material y métodos: Se ha revisado la bibliografía más reciente y se han comparado los resultados con la encuesta enviada a los 108 inscritos en la II Reunión Nacional de Nefrourología Pediátrica (nefrólogos y urólogos pediátricos, principalmente) sobre sus pautas de actuación. Se obtuvieron 30 respuestas.
Resultados: Casi el 90% de las hidronefrosis de diagnóstico intraútero son transitorias. Los pacientes con un diámetro anteroposterior de la pelvis renal en ecografía <15 mm, realizada no antes del tercer día de vida, no deben ser objeto de pruebas invasivas. No se recomienda realizar una cistografía a todos los niños con dilatación de manera sistemática. En el renograma diurético, la pérdida de función renal en sucesivos renogramas es el principal indicador de intervención. Se recomienda realizar profilaxis antibiótica en pacientes de riesgo.
Conclusión: Las respuestas a la encuesta coinciden mayoritariamente con lo recomendado en la bibliografía. El plan inicial con estos pacientes debe ser mínimamente invasivo. Los estudios deben realizarse en el momento del nacimiento en caso de sospecha de obstrucción bilateral o de vía común. En caso de dilataciones unilaterales, la evaluación debe efectuarse a partir del tercer día de vida. Recomendamos realizar profilaxis antibiótica, al menos hasta finalizar los estudios, en las hidronefrosis graves.
Supraglotitis atípica en la era postepiglotitis
Sr. Director:
La supraglotitis es una inflamación aguda de la epiglotis/supraglotis, de causa generalmente infecciosa, que suele provocar una obstrucción rápida y potencialmente letal de la vía respiratoria, por lo que se considera una emergencia médica1,2. Clásicamente, y de forma típica, era consecuencia de una infección por Haemophilus influenzae tipo b (Hib) que, tras la implantación en nuestro medio de la vacunación universal frente al mismo, ha pasado a ser una rareza y, por tanto, una entidad clínica casi desconocida para los pediatras con menos de 15 años de experiencia. La práctica erradicación del Hib como agente causal, condiciona un incremento relativo de otras causas2,3, tanto infecciosas (bacterianas, víricas o fúngicas) como no infecciosas (edema angioneurótico, traumatismos, lesiones cáusticas...), así como de otras posibilidades que por su curiosidad merecen ser reseñadas.
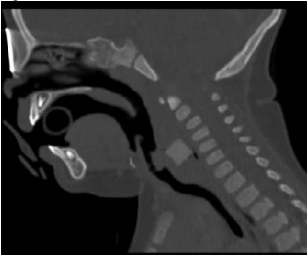 Figura 1. Tomografía computarizada. Corte sagitalPresentamos el caso de un niño de 18 meses de edad, remitido a la unidad de cuidados intensivos (UCI) de nuestro centro, con la sospecha clinicorradiológica (radiografía lateral de cuello) de epiglotitis aguda. El paciente carecía de antecedentes familiares o personales de interés y seguía el calendario de inmunizaciones vigente en nuestra comunidad autónoma, que incluye a esta edad 4 dosis de vacuna conjugada frente a Hib. La familia refería que el niño estaba previamente asintomático, excepto por cierta disminución de la ingesta en los días anteriores, y presentaba desde hacía 12 horas un cuadro de fiebre elevada (39,5 oC), decaimiento y dificultad respiratoria progresiva, con rechazo alimentario y babeo casi continuo. La exploración física revelaba una moderada afectación del estado general y fiebre (38,5 oC), posición en «trípode», babeo continuo, hipofonía con estridor inspiratorio leve e intermitente, sin tiraje intercostal significativo. La coloración cutánea y la hidratación de las mucosas eran normales. La auscultación cardiopulmonar era simétrica y mostraba una discreta hipoventilación bilateral. Los pulsos distales eran palpables, con una presión arterial en percentiles para la edad y el tiempo de relleno capilar normal. En los estudios complementarios realizados, la analítica mostraba los siguientes resultados: 11.740 leucitos/mm3 (72% neutrófilos), hemoglobina 9,7 g/dL, plaquetas 436.000/mm3, y proteína C reactiva 38,64 mg/L (valores normales: 0-3). La radiografía lateral de cuello revelaba una probable epiglotitis. Con el juicio clínico de epiglotitis aguda se inició tratamiento antibiótico con ceftriaxona y metilprednisolona. Para confirmar el diagnóstico y facilitar la intubación endotraqueal electiva del paciente se procedió a realizar una fibroendoscopia por vía nasal, que mostró una morfología normal de la epiglotis y la glotis. Ante la ausencia de hallazgos diagnósticos se realizó una tomografía computarizada de cuello con reconstrucción multiplanar, que puso de manifiesto la presencia de un cuerpo extraño (dado de parchís) entre la hipofaringe y la porción cervical del esófago, con un importante aumento de las partes blandas asociado, que provocaba una compresión laringotraqueal extrínseca significativa (figuras 1 y 2). El dado fue extraído mediante esofagoscopia sin incidencias, lo que propició una mejoría clínica rápida y progresiva del paciente. La familia no refirió en ningún momento, ni a priori ni a posteriori, sospecha de episodios de atragantamiento.
Figura 1. Tomografía computarizada. Corte sagitalPresentamos el caso de un niño de 18 meses de edad, remitido a la unidad de cuidados intensivos (UCI) de nuestro centro, con la sospecha clinicorradiológica (radiografía lateral de cuello) de epiglotitis aguda. El paciente carecía de antecedentes familiares o personales de interés y seguía el calendario de inmunizaciones vigente en nuestra comunidad autónoma, que incluye a esta edad 4 dosis de vacuna conjugada frente a Hib. La familia refería que el niño estaba previamente asintomático, excepto por cierta disminución de la ingesta en los días anteriores, y presentaba desde hacía 12 horas un cuadro de fiebre elevada (39,5 oC), decaimiento y dificultad respiratoria progresiva, con rechazo alimentario y babeo casi continuo. La exploración física revelaba una moderada afectación del estado general y fiebre (38,5 oC), posición en «trípode», babeo continuo, hipofonía con estridor inspiratorio leve e intermitente, sin tiraje intercostal significativo. La coloración cutánea y la hidratación de las mucosas eran normales. La auscultación cardiopulmonar era simétrica y mostraba una discreta hipoventilación bilateral. Los pulsos distales eran palpables, con una presión arterial en percentiles para la edad y el tiempo de relleno capilar normal. En los estudios complementarios realizados, la analítica mostraba los siguientes resultados: 11.740 leucitos/mm3 (72% neutrófilos), hemoglobina 9,7 g/dL, plaquetas 436.000/mm3, y proteína C reactiva 38,64 mg/L (valores normales: 0-3). La radiografía lateral de cuello revelaba una probable epiglotitis. Con el juicio clínico de epiglotitis aguda se inició tratamiento antibiótico con ceftriaxona y metilprednisolona. Para confirmar el diagnóstico y facilitar la intubación endotraqueal electiva del paciente se procedió a realizar una fibroendoscopia por vía nasal, que mostró una morfología normal de la epiglotis y la glotis. Ante la ausencia de hallazgos diagnósticos se realizó una tomografía computarizada de cuello con reconstrucción multiplanar, que puso de manifiesto la presencia de un cuerpo extraño (dado de parchís) entre la hipofaringe y la porción cervical del esófago, con un importante aumento de las partes blandas asociado, que provocaba una compresión laringotraqueal extrínseca significativa (figuras 1 y 2). El dado fue extraído mediante esofagoscopia sin incidencias, lo que propició una mejoría clínica rápida y progresiva del paciente. La familia no refirió en ningún momento, ni a priori ni a posteriori, sospecha de episodios de atragantamiento.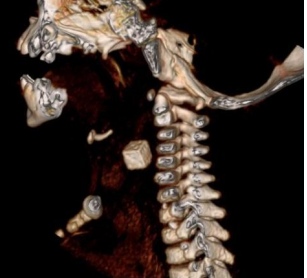 Figura 2. Tomografía computarizada. Reconstrucción multiplanar en 3 dimensionesEl caso presentado es destacable tanto por la clínica sugestiva de un proceso infeccioso agudo que provoca una obstrucción severa de la vía respiratoria superior (epiglotitis/supraglotitis) como por estar causado por un cuerpo extraño cúbico localizado en la zona faringoesofágica. La impactación de cuerpos extraños en esta zona es relativamente frecuente en niños4. La ausencia de un antecedente claro de atragantamiento, o de síntomas respiratorios al inicio (este paciente probablemente portaba el cuerpo extraño en su vía digestiva superior desde hacía varios días), no deben obviar esta posibilidad diagnóstica.
Figura 2. Tomografía computarizada. Reconstrucción multiplanar en 3 dimensionesEl caso presentado es destacable tanto por la clínica sugestiva de un proceso infeccioso agudo que provoca una obstrucción severa de la vía respiratoria superior (epiglotitis/supraglotitis) como por estar causado por un cuerpo extraño cúbico localizado en la zona faringoesofágica. La impactación de cuerpos extraños en esta zona es relativamente frecuente en niños4. La ausencia de un antecedente claro de atragantamiento, o de síntomas respiratorios al inicio (este paciente probablemente portaba el cuerpo extraño en su vía digestiva superior desde hacía varios días), no deben obviar esta posibilidad diagnóstica.
En los pacientes con clínica sugestiva de epiglotitis es imprescindible realizar un rápido diagnóstico de sospecha y establecer un adecuado manejo de la vía respiratoria para evitar complicaciones graves5. El diagnóstico es fundamentalmente clínico –el paciente presentaba todos los síntomas guía del cuadro–, y aunque puede complementarse con la radiografía simple de la vía respiratoria en proyección lateral, no se debe esperar de ella una suficiente sensibilidad ni especificidad6. Una vez sospechada, la epiglotitis se convierte en una emergencia médica que tiene como prioridad terapéutica el control de la vía respiratoria mediante intubación endotraqueal7. Dado que la intubación puede ser complicada, incluso en manos expertas, se recomienda utilizar dispositivos para el manejo de la vía respiratoria difícil, como la intubación guiada por fibroendoscopia, en un medio dotado con recursos humanos y materiales adecuados8 (UCI o quirófano).
En conclusión, la obstrucción aguda de la vía respiratoria superior en un lactante con signos de infección en la era posvacunación anti-Hib puede tener diversas causas, entre las que se encuentra la impactación faríngea de un cuerpo extraño. Con independencia de la causa, es prioritario asegurar la vía respiratoria por parte de profesionales con experiencia, habilidad y recursos técnicos suficientes en un entorno adecuado.
Bibliografía
1. Roosevelt G. Obstrucción inflamatoria aguda de las vías altas (crup, epiglotitis, laringitis y traqueítis bacteriana). En: Kliegman RM, Behrman RE, Jenson HB, Stanton BF, eds. Nelson textbook of pediatrics (ed. esp.). Madrid: Saunders-Elsevier España, 2009; 1.762-1.767.
2. Faden H. The dramatic change in the epidemiology of pediatric epiglotitis. Pediatr Emerg Care. 2006; 22(6): 443-444.
3. Sobol SE, Zapata S. Epiglottitis and croup. Otolaryngol Clin North Am. 2008; 41(3): 551-566.
4. McGahren ED. Esophageal foreign bodies. Pediatr Rev. 1999; 20(4): 129-133.
5. D'Agostino J. Pediatric airway nightmares. Emerg Med Clin North Am. 2010; 28(1): 119-126.
6. Dawson KP, Steinberg A, Capaldi N. The lateral radiograph of neck in laryngo-tracheo-bronchitis (croup). J Qual Clin Pract. 1994; 14(1): 39-43.
7. Rafei K, Lichenstein R. Airway infectious disease emergencies. Pediatr Clin North Am. 2006; 53(2): 215-242.
8. Damm M, Eckel HE, Jungehülsing M, Roth B. Airway endoscopy in the interdisciplinary management of acute epiglottitis. Int J Pediatr Otorhinolaryngol. 1996; 38(1): 41-51.
Hidrometrocolpos, polidactilia postaxial y anomalías cardiacas: el síndrome de McKusick-Kaufman como reto diagnóstico prenatal y posnatal
El síndrome de McKusick-Kaufman se caracteriza por la presencia de hidrometrocolpos, polidactilia postaxial y anomalías cardiacas en las mujeres, y malformaciones genitales en los varones. Mostramos el caso de una recién nacida que presentaba una gran masa quística intraabdominal e hidronefrosis bilateral en la ecografía practicada en la semana 32 de gestación. Tras el nacimiento, se confirmó dicha masa quística, así como la existencia de hidrometrocolpos, polidactilia postaxial y comunicación interauricular, por lo que fue diagnosticada de síndrome de McKusick-Kaufman.
Nefroma mesoblástico congénito con hiperecogenicidad medular que simula nefrocalcinosis
El nefroma mesoblástico congénito (NMC) es un tumor raro, siendo el más frecuente a nivel renal en los pacientes menores de 2 meses. Su origen histológico es la estroma renal inmadura, y se distinguen los subtipos clásico, mixto y celular. El tratamiento de elección es quirúrgico y su pronóstico es excelente. Se han descrito casos de NMC asociado a nefrocalcinosis en relación con la hipercalcemia paraneoplásica. Exponemos el caso de un recién nacido que presenta en la ecografía imágenes de hiperecogenicidad medular renal bilateral, similar a una nefrocalcinosis, en el contexto clínico de un NMC.
Convulsión hiponatrémica con parada respiratoria de etiología poco común
Las crisis convulsivas pueden ser la primera manifestación de una patología insospechada. Junto con el tratamiento anticonvulsivo, es necesario un protocolo de actuación dirigido a descartar y tratar las posibles causas reversibles. Las convulsiones por hiponatremia son una manifestación de máxima gravedad con claro riesgo vital, por lo que es necesaria una rápida actuación que eleve la natremia y la osmolaridad a cifras de seguridad. Una causa infrecuente de hiponatremia en los lactantes es la pérdida renal de sodio, secundaria a una infección del tracto urinario. Su origen parece deberse a una resistencia a la acción de la aldosterona en los túbulos renales.
Presentamos el caso clínico de un lactante de 49 días de vida, que presentó una crisis convulsiva y una parada respiratoria en el contexto de una hiponatremia grave (Na+: 110 mEq/L). El urocultivo confirmó una pielonefritis por Enterobacter aerogenes, y la ecografía una pielocaliectasia bilateral con cistografía normal, sin evidencia de reflujo vesiculoureteral.
Práctica clínica apropiada en el programa de cribado universal de hipoacusia en el recién nacido
Sr. Director:
El artículo de Sequí et al.1, publicado en Acta Pediátrica Española, expone la experiencia sobre el programa de cribado universal de hipoacusia en el recién nacido (RN) de un hospital comarcal de Valencia (3.309 RN en 3 años) y ofrece unos consejos para un modelo de organización más factible, eficiente y efectivo, avalado por la experiencia reconocida de su autor en esta línea de investigación. Creo que es un artículo honesto y útil, pero que merece una reflexión y comentarios de algunos puntos, avalados por pruebas científicas actuales, según las cuales no permiten establecer con claridad si el programa de cribado universal de hipoacusia en el RN facilita el desarrollo de una práctica clínica (en sus 3 componentes, científico-técnico, relacional-percibido y organizativo-económico) más apropiada que el cribado universal selectivo2-9.
• Hemos de confirmar que el programa cumple los objetivos en todos los niveles (primer nivel, cribado con otoemisiones acústicas; segundo nivel, confirmación mediante un segundo pase con otoemisiones; tercer nivel, diagnóstico mediante potenciales evocados; cuarto nivel, tratamiento; quinto nivel, evaluación). Como es habitual en este tipo de estudios8, el autor nos ofrece resultados «intermedios» hasta el tercer nivel (17 hipoacusias con déficit >30 dB HL, 5 de las cuales tenían factores de riesgo), sin ofrecernos resultados «finales» del tipo de tratamiento y evaluación a medio plazo (mejoría del lenguaje y comunicación en la edad preescolar) y a largo plazo (mejoría de la salud mental, función psicosocial y cognitiva, rendimiento escolar y ocupacional a lo largo de la vida), que es lo que marca la verdadera utilidad de todo programa en términos de calidad de vida relacionada con la salud. Se puede argumentar que ése no era el objetivo de su artículo, pero, en cualquier caso, sí se pretendía establecer el tipo de hipoacusia detectada, congénita bilateral moderada (41-60 dB HL), grave (61-80 dB HL) y/o profunda (>80 dB HL), y cuál de ellas se asocia con factores de riesgo. Al contabilizar las situaciones de hipoacusia leve (30-40 dB HL en este estudio) y/o unilateral estamos sobrestimando la verdadera utilidad de la prueba, pues consideramos como verdaderos positivos los casos con enfermedad poco significativa, a la hora de plantear el «punto crítico de irreversibilidad» en la etapa subclínica de la enfermedad, y el «tiempo de adelanto diagnóstico» de una prueba de cribado, frente al diagnóstico clínico usual en la etapa clínica, así como el verdadero valor a largo plazo de un diagnóstico y tratamiento precoz de la hipoacusia bilateral moderada-profunda9.
• Aun cuando el cribado universal puede ser más efectivo que el selectivo para la identificación de un porcentaje mayor de RN con hipoacusia, no se ha confirmado que tales programas aporten necesariamente mejores resultados a largo plazo, sobre la base de las revisiones sistemáticas existentes con ausencia de estudios de calidad bien controlados6,10. Algunas preguntas fundamentales son:
–¿Cuántos casos más se identifican con el cribado universal frente al selectivo? El NNT (número de pacientes que es necesario tratar) se calcula en 1.440 RN para detectar un caso adicional y en 2.401 para realizar un tratamiento precoz adicional (antes de los 10 meses)2,3.
–¿En qué medida la identificación temprana de la sordera del cribado universal sobre el selectivo propicia un tratamiento precoz y éste mejora los resultados a largo plazo de los RN con hipoacusia bilateral moderada a profunda? En vista de la ausencia de ensayos clínicos aleatorizados en esta área y de la dificultad de realizarlos, una opción es llevar a cabo estudios del tipo «antes y después», y que éstos eviten los errores aleatorios y sistemáticos (sesgos) habituales2,3,6,7,10: resultados primarios relevantes y validados en el área del lenguaje y la capacidad cognitiva, defectos metodológicos (tamaño muestral pequeño, no aleatorización de los grupos, no descripción adecuada de la población de estudio, no enmascaramiento de los grupos, seguimiento incompleto, etc.) y factores de confusión (edad al momento del diagnóstico, edad al inicio del tratamiento, cociente intelectual y comorbilidad diferentes en los grupos comparados, características de los patrones de referencia y tratamientos realizados, etc.). Estos errores son habituales en los estudios epidemiológicos sobre hipoacusia, incluso en los más persuasivos respecto a la bondad del programa11-13. Los factores principales que deciden la forma en que la sordera afecta al desarrollo de un niño son el grado de deficiencia auditiva y la edad a la que se diagnostica, si bien el reciente estudio de Wake et al.14 concluye que el peor pronóstico en el área del lenguaje a los 7-8 años se relaciona con las formas más graves de hipoacusia, pero no con el diagnóstico tardío.
Aunque Sequí et al.1 argumentan que las críticas efectuadas en la década de los noventa por los doctores Bess y Paradise (sobre la prueba y el programa de cribado de hipoacusia) están sobradamente contestadas por la comunidad científica, siento no compartir esta tranquilidad7-9, al igual que la US Preventive Service Task Force-Agency of Healthcare Research and Quality2,3 o la Cochrane Iberoamericana6.
• No es admisible argumentar las implicaciones legales para justificar la realización de un programa de cribado sin haber demostrado su verdadero alcance, y menos actualmente, en que las actividades preventivas se encuentran en debate permanente y se plantea que no siempre «hacer más es hacer mejor»15. Dado que se centran en el estudio de la población general sana, los programas preventivos deben estar fundamentados en el mayor nivel de evidencia científica para conocer los beneficios-perjuicios-costes, resaltando el potencial perjuicio del sobrediagnóstico (falsos positivos) y los sesgos del cribado (adelanto del diagnóstico, duración de la enfermedad y participación)9, lo que constituirá la base para establecer políticas de salud basadas en pruebas científicas, y no en la medicalización de la vida secundada por una medicina «proteccionista»16. Abogar por implicaciones legales me lleva a apoyar los comentarios de algunos autores, que, ante la «arrogancia» de la medicina preventiva, han llegado a preguntarse si no deberían llevar un etiquetado similar al que se ha impuesto en el tabaco, con «este programa de cribado puede afectar gravemente su salud»17,18... la de los pacientes y la de los médicos.
• En España la implantación del programa de cribado universal de hipoacusia es desigual; está establecido y en marcha en Asturias, Cantabria, Euskadi, Extremadura, La Rioja, Navarra y Valencia19. Sequí et al.1 comentan que en la Comunidad Valenciana el programa «cubre gran parte de los RN (datos no publicados)», y entendemos que la publicación de los datos empieza a ser prioritaria, pues no coincide con los datos de los hospitales de Alicante que cuentan con maternidad: el programa de cribado universal de hipoacusia está implantado en menos de la mitad de los 8 hospitales públicos y en ninguno de los 10 hospitales privados de la provincia. A esta falta de equidad se suma el distinto modelo organizativo de los distintos centros en los que se practica; por ejemplo, en mi hospital dista mucho de aproximarse al que proponen dichos autores.
• La decisión de desarrollar un programa de cribado precisa una evaluación externa e interna, en la que participen los tres actores básicos del sistema y los tres componentes de una práctica clínica apropiada en gestión clínica9:
–Los médicos pondrán el acento en la calidad científico-técnica (según los conceptos de eficacia, efectividad y seguridad), así como la facilidad de aplicación de la prueba y de ulteriores acciones (tratamientos); en esta valoración intervienen los resultados de los ensayos clínicos comunitarios y, eventualmente, los consensos científicos.
–Los pacientes pondrán el acento en la calidad relacional-percibida (según los conceptos de satisfacción, aceptabilidad e información).
–Las autoridades sanitarias pondrán el acento en la calidad organizativo-económica (según los conceptos de eficiencia, accesibilidad y equidad).
La excelencia médica intenta evitar errores y sesgos en el proceso asistencial médico; si es importante evitar los errores en el proceso diagnóstico-terapéutico individual, cuánto más lo será cuando este proceso atañe a la población, como es el caso de las pruebas de cribado. Richard Smith apuntaba, como editor de British Medical Journal, «cuando estoy enfermo quiero ser atendido por doctores que duden cada día del valor y la sensatez de lo que hacen». Y ése es el valor añadido del trabajo de Sequí et al.1 y de mi carta al director: permitir desarrollar una evidence-based medicine, evitando los riesgos para la ciencia y para los pacientes de la evidence-b(i)ased medicine20.
Bibliografía
1. Sequí Canet JM, Collar del Castillo J, Lorente Mayor L, Oller Prie¬to A, Morant Barber M, Peñalver Giner O, et al. Organización del cribado auditivo neonatal con otoemisiones en hospitales secundarios: factible, eficiente y efectivo. Acta Pediatr Esp. 2005; 63: 465-470.
2. US Preventive Service Task Force. Newborn hearing screening. Am Fam Physician. 2001; 64: 1.995-1.999.
3. Helfand M, Thompson D, Davis R, McPhillips H, Lieu TL, Homer CH. Newborn hearing screening [en línea] [citado 21 Ene 2006]. Disponible en: http://www.ahrq.gov/clinic/3rduspstf/newbornscreen/newborn sum1.htm#Comment
4. Kerschner JE. Neonatal hearing screening: to do or not to do? Pediatr Clin North Am. 2004; 51: 725-736.
5. Connolly JL, Carron JD, Roark SD. Universal newborn hearing screening: are we achieving the Joint Committee on Infant Hea¬ring (JCIH) objectives? Laryngoscope. 2005; 115: 232-236.
6. Puig T, Municio A, Medà C. Cribado (screening) auditivo neonatal universal versus cribado (screening) selectivo como parte del tratamiento de la sordera infantil (Revisión Cochrane traducida). En: La Biblioteca Cochrane Plus, 2005. Número 3. Oxford: Update Software Ltd. [en línea] [citado 21 Ene 2006]. Disponible en: http://www.Update-software.com (traducida de The Cochrane Library, 2005 Issue 3. Chichester: John Wiley&Sons, Ltd.).
7. González de Dios J, Mollar Maseres J, Rebagliato Russo M. Eva¬luación del programa de detección precoz universal de la hipoacusia en el recién nacido. An Pediatr (Barc). 2005; 63: 230-237.
8. González de Dios J, Mollar Maseres J. Cribado universal de hipoacusia neonatal: evaluación de la prueba frente a evaluación del programa. Acta Otorrinolaringol Esp. 2005; 56: 331-334.
9. González de Dios J, Mollar Maseres J, Rebagliato Russo M. Eva¬luación de las pruebas y programas de detección precoz (cribado o screening) de enfermedades. Rev Pediatr Aten Primaria. 2006 (en prensa).
10. Thompson D, McPhilips H, Davis R, Lieu T, Homer C, Helfand M. Universal newborn hearing screening: summary of evidence. JAMA. 2001; 286: 2.000-2.010.
11. Yoshinaga-Itano C, Sedey AL, Coulter DK, Mehl AL. Language of early and later-identified children with hearing loss. Pediatrics. 1998; 102: 1.161-1.171.
12. Moeller MP. Early intervention and language development in children who are deaf and hard of hearing. Pediatrics. 2000; 106: e43.
13. Kennedy C, McCann D, Campbell MJ, Kimm L, Thornton R. Universal newborn screening for permanent childhood hearing impairment: an 8-year follow-up of a controlled trial. Lancet. 2005; 366: 660-662.
14. Wake M, Poulakis Z, Hughes EK, Carey-Sargeant C, Rickards FW. Hearing impairment: a population study of age at diagnosis, seve¬rity, and language outcomes at 7-8 years. Arch Dis Child. 2005; 90: 238-244.
15. Godlee F. Preventive medicine make us miserable. BMJ. 2005; 330 [en línea] [citado 21 Ene 2006]. Disponible en: http://bmj.bmjjournals.com/cgi/reprint/330/7497/0-f
16. Márquez S, Meneu R. La medicalización de la vida y sus protagonistas. Gestión Clínica y Sanitaria. 2003; 5: 47-53 [en línea] [citado 21 Ene 2006]. Disponible en: http://www.iiss.es/gc/gestion16.pdf
17. Sackett DL. The arrogance of preventive medicine. CMAJ. 2002; 167 [en línea] [citado 21 Ene 2006]. Disponible en: http://www.cmj.ca/cgi/content/full/167/363
18. Stewart-Brown S, Farmer A. Screening could seriously damage your health. BMJ. 1997; 314: 533-534.
19. Alzina de Aguilar V. Detección precoz de la hipoacusia en el recién nacido. An Pediatr (Barc). 2005; 63: 193-198.
20. Peiró S. La construcción de la evidence b(i)ased medicine. Gestión Clínica y Sanitaria. 2005; 7: 131-138 [en línea] [citado 15 Ene 2006]. Disponible en: http://www.iiss.es/gc/gestion26.pdf
Síndrome de Van der Woude
El síndrome de Van der Woude se caracteriza por la presencia de hoyuelos (pits) o fístulas labiales, asociados a hendiduras labiales y/o palatinas. También se han asociado otras anomalías congénitas, como defectos cardiacos y anomalías en los miembros. Es un trastorno genético con herencia autosómica dominante, con penetrancia casi completa y expresividad variable, estando implicados dos locus génicos. En este artículo repasamos la prevalencia, la etiología y los aspectos clínicos característicos de este síndrome, además de presentar el caso de un recién nacido varón con labio leporino bilateral y apéndices mucosos en el labio inferior. Insistimos en la importancia de la asociación de las hendiduras faciales junto a los pits, por el patrón hereditario del síndrome, así como el diagnóstico diferencial de distintos cuadros clínicos que presentan hoyuelos mucosos.
Hernia discal lumbar en la infancia. Forma de presentación y características clínicas
Introducción: La hernia discal lumbar tiene escasa incidencia antes de los 21 años, siendo excepcional antes de los 10. La forma de presentación clínica es diferente a la del adulto, por lo que resulta difícil establecer el diagnóstico. Se revisa la bibliografía para conocer mejor las peculiaridades del cuadro clínico en el niño, diferenciarlas de las del adulto y aumentar el índice de sospecha diagnóstica.
Caso clínico: Niña de 9 años y 11 meses que consulta por cojera y dolor en el hueco poplíteo izquierdo, de un mes de evolución, con rigidez y escoliosis lumbar, y signo de Lasègue positivo bilateral. La resonancia magnética (RM) muestra una hernia discal en L5-S1.
Discusión y conclusiones: Es una enfermedad poco frecuente, de diagnóstico difícil y tardío. Debe sospecharse ante un paciente con dolor ciático, pero también ante un dolor en el hueco poplíteo, especialmente cuando exista un signo de Lasègue o una retracción o tensión dolorosa y persistente de los isquiotibiales. La alerta aumentará cuando se añada una escoliosis y una gran restricción de la movilidad lumbar, sin olvidar sospecharla ante una alteración de la marcha poco clara en su origen.
Trago accesorio
El trago accesorio se trata de un nódulo del color de la piel, sésil o pediculado, único o múltiple, unilateral o bilateral. Su localización clásica es la región preauricular, pero puede localizarse en la región mandibular o cervical. Se debe establecer el diagnóstico diferencial, fundamentalmente con quistes epidermoides, fístulas y fibromas. En ocasiones, aparece asociado a otras anomalías del desarrollo de los arcos branquiales, como el síndrome de Goldenhar. El tratamiento quirúrgico responde a razones estéticas y conlleva la escisión completa del cartílago asociado.
Cefaleas agudas recurrentes en la infancia y adolescencia: análisis crítico de los criterios diagnósticos de la migraña
Objetivo: Estudiar las características epidemiológicas y clínicas de la migraña y la cefalea tensional en la edad pediátrica y analizar la validez de los criterios utilizados en el diagnóstico de la migraña.
Pacientes y métodos: Se han revisado 300 historias de pacientes con cefaleas agudas recurrentes, recogiéndose datos epidemiológicos y clínicos y, en su caso, exámenes complementarios. Los criterios diagnósticos aplicados fueron los de la International Headache Society (IHS). Se han calculado la sensibilidad, la especificidad y el cociente de verosimilitud de los criterios de Vahlquist, Prensky y de la IHS para la migraña.
Resultados: El 98,3% de los casos eran cefaleas primarias: migraña (50%) o cefalea tensional (48,3%). El 32,7% de las migrañas tenían aura. La edad de inicio de la migraña era de 8,7 años, y de la cefalea tensional 9,7 años (p <0,05), sin diferencias entre sexos. No obstante, en la migraña con aura la edad de comienza (9,8 años) y la prevalencia del sexo femenino (63,3%) eran significativamente mayores (p <0,05). En la cefalea tensional había una mayor prevalencia (p <0,05) de sexo femenino, procedencia urbana y rendimiento escolar excelente; y en la migraña había mayor prevalencia (p <0,05) de antecedentes familiares. En la migraña el dolor era unilateral (44,4%) o bilateral (55,9%), pulsátil (87,5%), empeoraba con el ejercicio (68,8%), interrumpía la actividad diaria (65,3%), y se acompañaba de vómitos (71%) y fotofobia/sonofobia (67%). En la cefalea tensional era bilateral (81,8%), opresivo (85,3%), apenas empeoraba con el ejercicio (22,3%) o interrumpía la actividad diaria (12,1%) y, ocasionalmente, se acompañaba de vómitos (7,3%) o fotofobia/sonofobia (18,9%). El carácter pulsátil, los vómitos, la unilateralidad y la intensidad moderada-severa eran los ítems de mayor capacidad discriminatoria; los criterios de la IHS eran los de mayor validez diagnóstica. Los exámenes complementarios no modificaron el diagnóstico.
Conclusiones: La migraña y la cefalea tensional son las causas más frecuentes de cefaleas agudas recurrentes en la edad pediátrica, de inicio preferentemente en la edad escolar. Aunque los criterios de la IHS permiten su diagnóstico diferencial, el control evolutivo sería la prueba de referencia para validar los criterios diagnósticos.
Deleción intersticial del brazo largo del cromosoma 4(4q12q21.1): comunicación de un nuevo caso
Se comunica un nuevo caso de deleción proximal del brazo largo del cromosoma 4 de novo, en un niño de 3 años de edad con rasgos fenotípicos compatibles con un síndrome de Waardemburg tipo II. Presentaba mechón de pelo blanco frontal, hipoacusia neurosensorial bilateral, desplazamiento lateral de cantos internos, heterocromía de iris, fisura velopalatina, lesiones hipocrómicas en tronco, hipotonía axial, extremidades cortas, deformidades de cuerpos vertebrales, retraso mental y ponderoestatural, reflujo gastroesofágico, síndrome de malabsorción, panhipopituitarismo, comunicación interauricular tipo ostium secundum, hipermetropía (11 dioptrías) y dificultad para la deglución. El cariotipo de alta resolución realizado en células de sangre periférica y piel hipo/hiperpigmentada puso de manifiesto una deleción intersticial en el brazo largo del cromosoma 4(4q12-q21.1). El estudio mutacional del gen MITF (Waardenburg II) fue normal.
Se revisan los casos similares descritos anteriormente en la bibliografía y se resalta que la asociación retraso mental y ponderoestatural en niños con rasgos fenotípicos que recuerdan al síndrome de Waardenburg o al piebaldismo aislado deben alertar sobre posibles deleciones en la estructura del brazo largo del cromosoma 4.
Nevo comedoniano. Respuesta satisfactoria al ácido retinoico
Sr. Director:
El nevo comedoniano es una malformación folicular constituida por múltiples lesiones con aspecto de comedones de distintos tamaños, agrupados linealmente o en parches, que se distribuyen habitualmente de forma unilateral, aunque se han descrito algunos casos de distribución bilateral1.
Un niño de 6 años, sin antecedentes de interés, acude a nuestra consulta por presentar desde hace un año y medio unas lesiones en la zona posterolateral derecha del cuello, ocasionalmente pruriginosas.
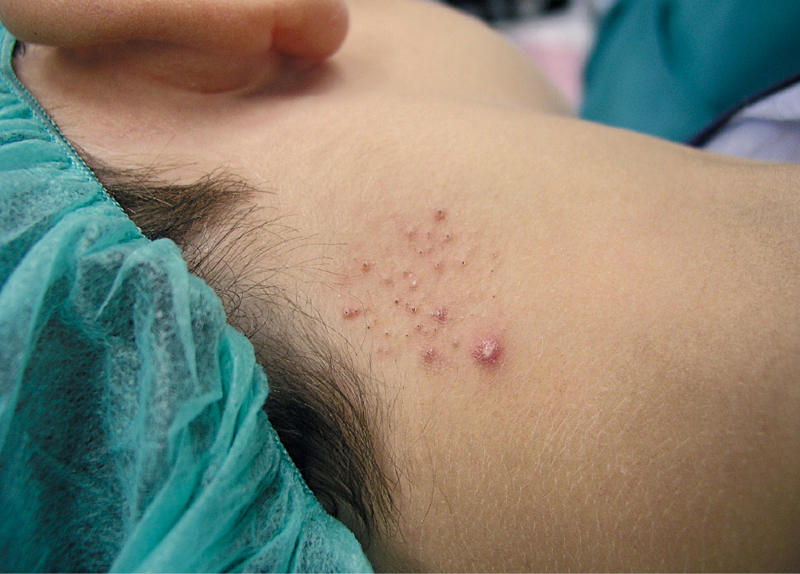
En la exploración dermatológica se observan, sobre un área bien delimitada de aproximadamente unos 3 cm de diámetro, múltiples pápulas centradas por tapones hiperqueratósicos similares a comedones abiertos, con alguna pústula aislada (figura 1).
No presenta antecedentes familiares de lesiones similares.
Ante la sospecha de nevo comedoniano, se realizó una biopsia que confirmó dicho diagnóstico.
Se inició tratamiento con ácido retinoico al 0,025% una vez al día, desapareciendo gran parte de las lesiones en aproximadamente 2 meses.
El nevo comedoniano puede aparecer en la pubertad o estar presente desde el nacimiento. Suelen ser lesiones asintomáticas y generalmente unilaterales, con preferencia por ciertas zonas, como la cara, el cuello, el tronco o las extremidades superiores1,2. Su tamaño es variable y oscila desde unos pocos centímetros a lesiones extensas que afectan a un hemicuerpo entero2.
Su etiopatogenia no está clara, pero parece tratarse de un mosaicismo genético2.
Es necesario realizar un diagnóstico diferencial con la acné vulgar y neonatal. La distribución unilateral y la persistencia del nevo comedoniano lo diferencian de estas entidades. Otras afecciones con las que se puede confundir son la cloracné, los comedones disqueratósicos familiares, el nevo ostial poroqueratósico ecrino, la enfermedad de Darier lineal y el nevo sebáceo2.
Dado que es una lesión benigna no necesita tratamiento, salvo por motivos estéticos o para evitar infecciones secundarias.
La extirpación quirúrgica es una opción terapéutica en las lesiones localizadas y de escasa extensión2.
Otros tratamientos que se pueden valorar, aunque con respuesta variable, son los siguientes: láser de CO2, dermoabrasión, derivados de la vitamina D3 tópicos (calcipotriol, tacalcitol)3, agentes queratolíticos, como el ácido salicílico, los hidroxiácidos alfa y las cremas de lactato amónico al 12%4. Otra opción, que en el paciente del caso expuesto tuvo una respuesta muy satisfactoria, es el ácido retinoico5 tópico, en sus distintas concentraciones.
Bibliografía
- Izquierdo MJ, Requena C, Requena L. Nevo comedoniano. En: Neoplasias anexiales cutáneas. Madrid: Grupo Aula Médica, 2004; 211-214.
- Monteagudo Sánchez B, Ginarte Val M, León Muiños E, Vázquez Golpe R, Varela Iglesas A. Nevo comedoniano. An Pediatr (Barc). 2006; 65(2): 171-172.
- Wakahara M, Kiyohara T, Kumakiri M, Kuwahara H, Fujita T. Bilateral nevus comedonicus: efficacy of topical tacalcitol ointment. Acta Derm Venereol. 2003; 83: 51.
- Bordel MT, Miranda A. Nevo comedoniano unilateral: eficacia tras el tratamiento con lactato amónico al 12%. Actas Dermosifiliogr. 2006; 97: 150.
- Decherd JW, Mills O, Leyden JJ. Naevus comedonicus treatment with retinoic acid. Br J Dermatol. 1972; 86: 528-529.
Talón negro (petequias del calcáneo)
El talón negro (petequias del calcáneo) es una lesión asintomática, de etiología traumática, benigna y autolimitada, que suele afectar a adolescentes y adultos jóvenes deportistas. Su localización más frecuente es en la parte posterior o posterolateral de uno o ambos talones. Se trata de la expresión clínica de una hemorragia dérmica con eliminación transepidérmica (estrato córneo). Su evolución habitual es la completa desaparición si cesa la actividad causal, en muchos casos el deporte. Por su carácter asintomático y benigno, no precisa tratamiento. Su importancia radica en el crucial diagnóstico diferencial que se establece con las lesiones pigmentadas, entre ellas con el melanoma maligno. La localización de la lesión, su bilateralidad, la ausencia de síntomas y su aparición en un joven deportista son importantes claves diagnósticas, que pueden permitir establecer un diagnóstico correcto y evitar una biopsia cutánea innecesaria.
La esquisencefalia: malformación cerebral infrecuente
Introducción: La esquisencefalia (ESQ) es un raro trastorno de la migración neuronal que se caracteriza por la presencia de hendiduras que, atravesando el hemisferio cerebral, se extienden desde los ventrículos laterales hasta la superficie cortical.
Caso clínico: Mujer de 11 años con discapacidad intelectual y crisis convulsivas parciales que progresan hacia un síndrome de Lennox-Gastaut de difícil control.
La resonancia magnética (RM) cerebral muestra una ESQ parietooccipital bilateral de labio abierto en el lado derecho.
Discusión: Clínicamente, la ESQ se caracteriza por diversas discapacidades del desarrollo, entre las que destacan la hemiparesia y las crisis convulsivas. La RM es la modalidad de imagen de elección para el diagnóstico de ESQ. La gravedad del trastorno motor se ve muy influenciada en función de la extensión, la apertura labial, la bilateralidad y el tamaño de la malformación.
Encefalocele adquirido en un paciente con enfermedad de Albers-Schönberg
La osteopetrosis es una rara enfermedad ósea caracterizada por una esclerosis del esqueleto y causada por un defecto en la resorción ósea por parte de los osteoclastos. La forma autosómica dominante de la osteopetrosis se divide en 2 subtipos. El tipo I implica un notable engrosamiento de la bóveda craneal. En el tipo II, o enfermedad de Albers-Schönberg, predomina la esclerosis vertebral y de la base del cráneo.
Las complicaciones más frecuentemente descritas de la osteopetrosis se localizan en el sistema nervioso, secundarias a la compresión de los pares craneales, los vasos sanguíneos y la médula espinal, por la oclusión gradual o una falta de desarrollo de los orificios craneales.
Se presenta el caso de un niño de 5 años, cuya enfermedad se inició con una proptosis ocular bilateral de aparición brusca, cefalea y vómitos, secundaria a un encefalocele producido por una fractura espontánea del techo de la órbita.
Criptorquidia y otras anomalías del descenso testicular
La criptorquidia o testículo no descendido es el trastorno de las glándulas endocrinas masculinas más común en los niños. Se define como la falta de testículo en el escroto, secundaria a una anomalía en el proceso de descenso, y se engloba en el síndrome de escroto vacío. Su incidencia varía entre el 3,4 y el 5,8% en los niños nacidos a término.
Las razones más importantes para su tratamiento son: fecundidad disminuida, aumento de la tasa de neoplasias malignas, riesgo aumentado de torsión testicular o de lesión contra el pubis y estigma psicológico del escroto vacío.
La criptorquidia es una manifestación bien conocida de anomalías cromosómicas y un componente común de más de 50 síndromes de anomalías congénitas múltiples.
El diagnóstico se realiza por la anamnesis y el examen físico debiendo determinar situación, tamaño, comparación con el otro lado, desarrollo del escroto, movilidad, presencia o no de reflejo cremastérico, tamaño y morfología del escroto y del pene y signos de hernia inguinal.
Puede clasificarse por su ubicación en teste no palpable o palpable, que, a su vez, se diferencian en criptorquidia o ectopia testicular.
En los casos de testes no descendidos palpables no es necesaria una evaluación analítica para contribuir al diagnóstico.
La hormonoterapia se realiza con gonadotropina coriónica (hCG) y está recomendada en testículos inguinales distales o en aquellos que se encuentren en la entrada del escroto.
En el caso de testes palpables uni- o bilateralmente se realiza una orquidopexia; en el caso de los no palpables se recomienda una incisión inguinal inicial para la exploración adecuada del canal inguinal: si se aprecia un testículo de características normales, se realiza orquidopexia; si se localizan testes atróficos o restos testiculares, se realiza exéresis; si no se localizan restos en el canal inguinal, se realiza una inspección intrabdominal con la óptica de laparoscopia. Actualmente, se recomienda para el tratamiento del testículo intrabdominal la orquidopexia asistida por laparoscopia.
Tratamiento de la incontinencia salival en el niño con patología neurológica
La incontinencia salival, o babeo, es un problema frecuentemente asociado a varios cuadros neuropediátricos. En los casos severos conlleva graves consecuencias clínicas y psicosociales. Se han propuesto múltiples procedimientos para su tratamiento, con resultados variables. El primer escalón terapéutico consiste en la combinación de entrenamiento logopédico o neurorrehabilitador y la aplicación de fármacos anticolinérgicos, entre los cuales los más utilizados son el trihexifenidilo y la escopolamina, este último aplicado mediante parches transdérmicos. Dados los efectos indeseables a distancia de estos fármacos, se comienza a utilizar la infiltración de las glándulas submaxilares con toxina botulínica A, bajo control ecográfico. Este procedimiento es efectivo en el 50% de los casos, pero presenta el inconveniente de que su efecto es temporal, y se requiere repetir la infiltración cada 4-6 meses. Finalmente, en pacientes con babeo invalidante que no responden a la terapia farmacológica y rehabilitadora, a largo plazo estaría indicada la realización de una técnica supresora definitiva. En la actualidad la técnica más segura y eficaz es la combinación de una submaxilectomía bilateral y la ligadura de ambos conductos parotídeos. Este sencillo procedimiento elimina de forma completa la incontinencia salival en más del 90% de los casos, con mínimas complicaciones, y debe seguirse de una correcta higiene bucodental.
Uveítis por cotrimoxazol
Niño de 10 años de edad que presenta hiperemia conjuntival bilateral, dolor ocular y visión borrosa tras la administración de trimetoprim-sulfametoxazol (TMP-SMX) por vía oral.
Análisis del programa de detección precoz de la hipoacusia neonatal en nuestro centro
Objetivos: Conocer los resultados del programa de detección precoz de hipoacusia en recién nacidos (RN) de nuestro hospital durante 2001 y 2004.
Pacientes y métodos: Se realizaron otoemisiones acústicas (OEA) a 2.461 (2001) y 2.549 RN (2004); si éstas eran negativas, se repetían antes de un mes. Los RN con la segunda otoemisión negativa eran remitidos al servicio de otorrinolaringología (ORL), completándose su valoración mediante potenciales auditivos troncoencefálicos.
Resultados: El 10% (n= 260) y el 5% (n= 145), respectivamente, no pasaron la primera OEA. Continuaron esta primera fase 234 (2001) y 144 (2004), y no pasaron la segunda OEA el 16% (n= 37) y el 24% (n= 35), generándose una remisión al servicio de ORL del 1,51 y el 1,37% cada año; 29 niños no completaron la fase diagnóstica. De los 72 RN con sospecha de hipoacusia, el 59,72% (n= 43) completó el programa, el 30,23% había estado ingresado, el 23,25% presentaba factores de riesgo y el 48,83% tenía OEA negativas bilaterales. En el servicio de ORL se confirmó la hipoacusia en 23 RN (53,48%): en un 57,14% era neurosensorial (un 62% en RN varones sin factores de riesgo; un 75% bilaterales y un 50% profundas) y en un 42,16% era de transmisión.
Conclusiones: Nuestra incidencia global de hipoacusia neurosensorial y de transmisión es de 4,54/1.000 RN, y para la neurosensorial profunda de 1,87/1.000 RN. El 71% de los hipoacúsicos eran RN sanos y sin factores de riesgo, predominando los varones. La capacidad de detección de las OEA fue significativamente mayor en 2004; las remisiones a ORL fueron adecuadas, las pérdidas de seguimiento en la primera fase escasas, pero elevadas en la de confirmación.
Malformación cerebral en dos niños inmigrantes
La esquisencefalia es un infrecuente trastorno de la migración neuronal, caracterizado por presentar una hendidura profunda cerebral, unilateral o bilateral, desde la piamadre hasta la superficie del ventrículo, recubierta de sustancia gris. Hay dos tipos: el tipo I, de labios cerrados, y el tipo II, más infrecuente, de labios abiertos. Suele cursar de forma asintomática en el neonato, y la clínica aparece en el segundo trimestre de la vida, con alteraciones en el perímetro craneal, espasmos infantiles, epilepsia parcial rebelde, monoparesia espástica, retraso mental con espasticidad, etc. Describimos los casos de dos pacientes inmigrantes diagnosticados de parálisis cerebral en sus países de origen, en los que se diagnosticaron los dos tipos de esquisencefalia al ser remitidos a nuestro centro. También pretendemos subrayar la rareza del tipo II.
Plagiocefalia posterior frente a aplanamiento occipital: diagnóstico y pautas terapéuticas
Introducción: Uno de los motivos de consulta relacionados con la morfología del cráneo cada vez más frecuentes en neuropediatría es el aplanamiento occipital unilateral o bilateral de la cabeza del niño. Es importante distinguir entre la plagiocefalia posterior verdadera por el cierre de la sutura lambdoidea y el aplanamiento occipital sin cierre de suturas, a fin de orientar sobre las medidas terapéuticas más adecuadas en cada caso.
Objetivo: Valorar la existencia o no de plagiocefalia posterior verdadera en niños con aplanamiento occipital.
Pacientes y métodos: Durante 4 años acudieron a la consulta un total de 42 niños menores de un año con problemas relacionados con la cabeza, 9 de los cuales presentaron un aplanamiento occipital. En los 9 casos se solicitó una tomografía computarizada craneal, y se recomendó el tratamiento más oportuno en cada paciente.
Resultados: Sólo un caso presentó una plagiocefalia posterior. A todos se les recomendó adoptar medidas posturales y realizar fisioterapia, y a tres niños se les colocó un casco. La remodelación del cráneo fue adecuada y tanto los niños como las familias toleraron el casco.
Conclusiones: La mayoría de los niños con aplanamiento occipital no presentan una plagiocefalia, por lo que las medidas posturales, la fisioterapia y, en algunos casos, la colocación de un casco son suficientes para mejorar e incluso normalizar la morfología craneal de los lactantes.
Importancia de la resonancia magnética en el síndrome del acueducto vestibular dilatado en la infancia
El síndrome del acueducto vestibular dilatado es la patología de herencia autosómica recesiva en la que se produce una hipoacusia progresiva o fluctuante desde la infancia por detención del desarrollo del oído interno, lo que provoca un aumento del saco endolinfático y una ligera displasia coclear.
Presentamos 2 casos clínicos cuya peculiaridad es que ambos son unilaterales, ya que suelen ser bilaterales en el 90% de los casos.
Una tomografía computarizada (TC) de cortes finos (1 mm) identifica al acueducto vestibular aumentado, pero una TC normal no excluye este diagnóstico, por lo que se debe realizar una resonancia magnética a todo paciente con hipoacusia neurosensorial unilateral para descartar ésta y otras alteraciones que pueden estar asociadas.
Pezón supernumerario: ¿un marcador cutáneo de anomalías hematológicas, cardiovasculares y renales?
Sr. Director:
 Los pezones supernumerarios (PS), también denominados pezones accesorios o politelia, son una anomalía congénita menor, relativamente común, y constituyen la patología mamaria accesoria más frecuente (tabla 1). Representan restos de las crestas mamarias embriológicas, engrosamientos ectodérmicos simétricos que se extienden desde la axila hasta la ingle. Para algunos autores, son un ejemplo de atavismo o aparición espontánea de características ancestrales en los miembros de una especie. Antiguamente se asociaban con la fertilidad, y en la época medieval se consideraron una marca del diablo1.
Los pezones supernumerarios (PS), también denominados pezones accesorios o politelia, son una anomalía congénita menor, relativamente común, y constituyen la patología mamaria accesoria más frecuente (tabla 1). Representan restos de las crestas mamarias embriológicas, engrosamientos ectodérmicos simétricos que se extienden desde la axila hasta la ingle. Para algunos autores, son un ejemplo de atavismo o aparición espontánea de características ancestrales en los miembros de una especie. Antiguamente se asociaban con la fertilidad, y en la época medieval se consideraron una marca del diablo1.
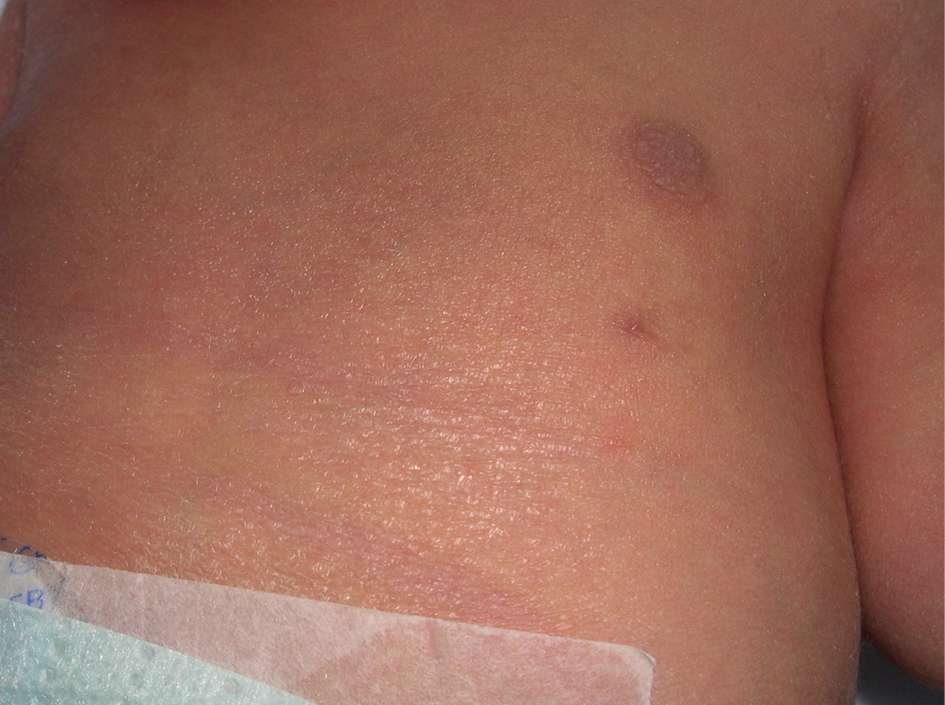
Según las series, su prevalencia varía desde el 0,22 hasta el 6%, son más frecuentes en los sujetos de raza negra, asiáticos, indígenas americanos, árabes y judíos que en los europeos de raza caucásica. No hay diferencias según el sexo, aunque se detecta un ligero predominio en los varones. Normalmente es una anomalía esporádica, aunque el 6-10% de los casos son familiares (herencia autosómica dominante con penetrancia incompleta o dominante ligada al cromosoma X). Los PS se localizan habitualmente en la región inframamaria, sobre todo la izquierda (figura 1). Pueden presentarse en la región supramamaria o en cualquier otra zona de las líneas mamarias embriológicas. En ocasiones se sitúan fuera de esas líneas, como la espalda, los hombros, la cara posterior de los muslos, la cara, el cuello o la vulva. Las lesiones suelen ser solitarias, pero hay casos múltiples (incluso 8) unilaterales o bilaterales. Clínicamente, se manifiestan como tumores pediculados pequeños, blandos y rosados o marrones. En el recién nacido las lesiones pueden ser muy tenues, en forma de máculas de 1-3 mm de tamaño y de color marrón claro2.
Generalmente, el diagnóstico se basa en la clínica y en su presencia desde el nacimiento. Pueden confirmarse mediante dermatoscopia, nuevas técnicas, como la microscopia confocal de reflexión, o el estudio histopatológico. Éste muestra el engrosamiento epidérmico, las estructuras pilosebáceas y el músculo liso, con o sin glándulas mamarias verdaderas. El diagnóstico diferencial se realiza con el nevo melanocítico, el neurofibroma, las cicatrices de amniocentesis y el pólipo anexial neonatal. Los PS son un proceso benigno, pero pueden desarrollar cualquier enfermedad que aparezca en las mamas normales, incluidos los tumores benignos y malignos. Normalmente no precisa tratamiento, pero en casos sintomáticos, o por motivos estéticos, pueden extirparse1,3.
Los PS se asocian a varios síndromes polimalformativos, como el síndrome de Simpson-Golabi-Behmel (trastorno de herencia recesiva ligada al cromosoma X caracterizado por sobrecrecimiento prenatal y posnatal, alteraciones craneofaciales, anomalías congénitas cardiacas, renales y/o esqueléticas y tumores embrionarios), varios subgrupos del síndrome de displasia ectodérmica, el síndrome 3-M, el síndrome de Killian/Teschler-Nicola y la disostosis espondilocostal. También se ha observado una coexistencia con el nevo de Becker o la lentiginosis unilateral parcial1.
Lo más importante de los PS es su posible relación con algunas enfermedades y, por tanto, su posible utilidad como marcador cutáneo. Aunque esta relación es controvertida4, recientemente han aparecido nuevos estudios que la apoyan5-7. Entre dichas enfermedades se incluyen las hematológicas (deficiencias de factores y leucemias agudas)5, las cardiovasculares6,8, el cáncer renal o genital9 y las malformaciones renales y de las vías urinarias7,10. Algunos autores explican esta última asociación por un desarrollo embriológico paralelo del sistema genitourinario y mamario. Mientras que algunos autores recomiendan realizar en todos los pacientes con PS7 un estudio renal, especialmente mediante ecografía, otros lo reservan para los casos familiares10 o los asociados a otras malformaciones3.
Bibliografía
- Antaya R, Schaffer JV. Anomalías del desarrollo. En: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatología, 1.ª ed. Madrid: Elsevier España, S.A.; 2004; 915-931.
- Brown J, Schwartz RA. Supernumerary nipples: an overview. Cutis. 2003; 71: 344-346.
- Merlob P. Congenital malformations and developmental changes of the breast: a neonatological view. J Pediatr Endocrinol Metab. 2003; 16: 471-485.
- Grotto I, Browner-Elhanan K, Mimouni D, Varsano I, Cohen HA, Mimouni M. Occurrence of supernumerary nipples in children with kidney and urinary tract malformations. Pediatr Dermatol. 2001; 18: 291-294.
- Aslan D, Gürsel T, Kaya Z. Supernumerary nipples in children with hematologic disorders. Pediatr Hematol Oncol. 2004; 21: 461-463.
- Rajaratnam K, Kumar PD, Sahasranam KV. Supernumerary nipple as a cutaneous marker of mitral valve prolapse in Asian Indians. Am J Cardiol. 2000; 86: 695-697.
- Ferrara P, Giorgio V, Vitelli O, Gatto A, Romano V, Bufalo FD, et al. Polythelia: still a marker of urinary tract anomalies in children? Scand J Urol Nephrol. 2009; 43: 47-50.
- Urbani CE. Supernumerary nipple and cardiocutaneous associations. J Am Acad Dermatol. 2004; 50: e9.
- Urbani CE, Betti R. Aberrant mammary tissue and nephrourinary malignancy. Cancer Genet Cytogenet. 1996; 87: 88-89.
- Brown J, Schwartz RA. Supernumerary nipples and renal malformations: a family study. J Cutan Med Surg. 2004; 8: 170-172.
Quilotórax congénito bilateral: descripción y manejo
El quilotórax constituye la causa más frecuente de derrame pleural en el recién nacido. Generalmente, se trata de un derrame unilateral y secundario a la cirugía torácica; en muy pocos casos es bilateral y de origen congénito.
Presentamos el caso clínico de un neonato con diagnóstico ecográfico prenatal de derrame pleural bilateral.
En la actualidad, el tratamiento para el manejo del quilotórax congénito no está claramente definido.
Valores normales de hormona antimülleriana en niños españoles
Introducción: La hormona antimülleriana (AMH) es el factor testicular responsable de la regresión de las estructuras derivadas de los conductos de Müller. En este trabajo se presentan los valores normales de la concentración de AMH en los varones españoles en edad pediátrica. Asimismo, se ofrece un gráfico de distribución en percentiles de la concentración de AMH, que puede resultar útil para conseguir diagnósticos precisos en la evaluación diagnóstica de gónadas bilateralmente no palpables y en los procesos intersexuales.
Pacientes y métodos: Se obtuvieron 240 muestras de sangre de niños de 0-18 años de edad, que fueron remitidos a nuestro servicio por diversas causas; 35 de las muestras se obtuvieron de sangre de cordón umbilical y se utilizaron para determinar la concentración de la hormona en el periodo neonatal. Se determinó la concentración por medio de radioinmunoanálisis por adsorción de enzimas usando anticuerpos contra la hormona recombinante humana.
Resultados: La concentración de AMH (en ng/mL) muestra un valor mínimo (desviación estándar) en el periodo neonatal de 31,90 (21,46), durante el cual no se encontraron diferencias estadísticamente significativas entre individuos de este grupo según su edad gestacional o su peso al nacimiento; entre el mes de vida y los 4 años se observa un aumento de la concentración (51,55 [30,36]), seguido de un periodo en meseta hasta los 8 años (50,85 [36,77]). Posteriormente, se aprecia un descenso hasta un mínimo a los 12 años (38,60 [28,92]), con una disminución hasta niveles basales a los 14 años (14,50 [16,65]), que se mantiene tras esta edad (12,05 [28,43]).
Conclusiones: Los resultados de este estudio coinciden con los de otros autores y confirman que la AMH puede utilizarse como marcador específico de tejido testicular en niños, especialmente en los neonatos, utilizando suero obtenido de sangre de cordón umbilical.
Parotiditis aguda supurativa neonatal
Sr. Director:
La parotiditis aguda supurativa es una enfermedad muy rara en el periodo neonatal (3.8/10.000 ingresos). Su diagnóstico es fácil, fundamentalmente clínico, cura sin repercusiones y son excepcionales las recidivas, al contrario de lo que ocurre en edades posteriores1,2.
Se da más en prematuros (38%)1, recién nacidos de bajo peso3 y varones (3:1)1. La infección asciende a través del conducto de Stenon, propiciada por ciertos factores que hacen que la saliva sea más espesa, como la deshidratación. De ahí que los prematuros con una mayor predisposición a la deshidratación tengan un riesgo incrementado.
Según otros autores, predomina la vía hematógena. En presencia de una septicemia, la parotiditis está propiciada por anomalías de la glándula, traumatismos orales, sonda orogástrica o inmunosupresión1-4. A veces es bilateral, en cuyo caso hay que descartar abscesos mamarios si el bebé está alimentado al pecho5.
Para el diagnóstico basta con observar si hay signos inflamatorios locales, secreción purulenta a través del conducto de Stenon al comprimir la glándula (patognomónico) y crecimiento de un germen patógeno en dicho material1-3. Rara vez se afecta la glándula salival submandibular, cuya secreción es más rica en IgA y lisozimas6. Los hallazgos de laboratorio son inespecíficos, salvo la hiperamilasemia, muy rara en los neonatos por la inmadurez enzimática1.
Debe realizarse un diagnóstico diferencial, basado en hallazgos ecográficos, con diversas entidades: celulitis, adenopatías, abscesos, miositis, hemangiomas, adenomas y lipomas. En particular, hay que descartar la celulitis-adenitis por Streptococcus agalactiae, forma poco habitual de sepsis neonatal tardía pero que puede ser muy grave7.
El germen más frecuentemente hallado es Staphylococcus aureus (55%), frente a estreptococos (22%).
El tratamiento se iniciará empíricamente, con una combinación de penicilina resistente a betalactamasas o cefalosporinas y aminoglucósidos. Menos del 25% de los casos precisan un drenaje de la glándula1-3,8,9.
Se expone el caso de un neonato de sexo femenino de 12 días de vida, que presentaba una tumefacción preauricular izquierda de unas horas de evolución, febrícula de 37,7 ºC y rechazo de las tomas. No tenía antecedentes personales de interés, salvo una rotura prematura de membranas de 13 horas, por lo que se administró a la madre una dosis de ampicilina (exudado rectovaginal materno para S. agalactiae negativo). Tomaba lactancia materna exclusiva. La madre estaba sana, sin lesiones mamarias.
Se determinó una temperatura de 37,4 ºC y la exploración por aparatos fue normal, salvo por la presencia de una tumefacción preauricular izquierda de 3 x 2 cm, dura y delimitada, con piel suprayacente normal, y un leve despegamiento del pabellón auricular izquierdo.
En las pruebas complementarias se detectaron los siguientes parámetros: leucocitosis con neutrofilia y trombocitosis (leucocitos 26,70 10E3/µL, 76% neutrófilos 20,29 10E3/µL, plaquetas 524.000/µL), alfa-amilasa <30 U/L y proteína C reactiva 13,2 mg/L.
En la ecografía cervical se observaba en la región preauricular izquierda, coincidiendo con la tumefacción palpable, un marcado aumento de espesor del tejido celular subcutáneo periparotídeo, con aspecto hiperecogénico homogéneo, así como un discreto aumento global del volumen de la glándula parótida izquierda. La ecogenicidad glandular era homogénea, sin adenopatías intraglandulares, ni dilatación de los conductos.
En el cultivo del material purulento que se obtuvo a través del conducto de Stenon, al comprimir la glándula, se aisló S. aureus.
Ante la sospecha clínica de parotiditis supurativa, se instauró tratamiento antibiótico empírico con cloxacilina y gentamicina intravenosa durante 10 días. La paciente presentó un pico febril de 38,6 ºC en las primeras 24 horas del ingreso, y posteriormente una febrícula durante 48 horas más. Se produjo una mejora progresiva de la tumefacción y de los signos inflamatorios locales hasta su completa desaparición.
El caso presentado pretende recordar a los pediatras la existencia de una patología poco frecuente en el periodo neonatal. Conviene tenerla en cuenta, en primer lugar, por sus diferencias con los niños mayores (ausencia de hiperamilasemia y etiología bacteriana) y, en segundo lugar, por la necesidad del diagnóstico diferencial con la celulitis-adenitis por S. agalactiae.
Bibliografía
- Spiegel R, Miron D, Sakran W, Horovitz Y. Acute neonatal suppurative parotitis: case reports and review. Pediatr Infect Dis J. 2004; 23(1): 76-78.
- Sabatino G, Verrotti A, De Martino M, Fussilli P, Pallota R, Chiarelli F. Neonatal suppurative parotitis: a study of five cases. Eur J Pediatr. 1999; 158: 312-314.
- Ayala Curiel J, Galán del Río P, Poza del Val C, Aguirre Conde A, Cotero Lavín A. Parotiditis aguda supurativa neonatal. An Pediatr (Barc). 2004; 60(3): 274-277.
- Martos Martínez D, Rodríguez Becerra A, Cintado Bueno C. Parotiditis supurada en el periodo neonatal. Aportación de dos casos y revisión de la literatura. An Esp Pediatr. 1998; 48: 415-416.
- Singh K. Bilateral parotid abscess in a neonate. Indian Pediatrics. 2006; 43(17): 1.009-1.010.
- Schwab J, Baroody F. Neonatal suppurative parotitis: a case report. Clin Pediatr. 2003; 42: 565-566.
- Soler Palacín P, Monfort Gil R, Castells Vilella L, Pagone Tangorra F, Serres i Créixams X, Balcells Ramírez J. Síndrome de celulitis-adenitis por estreptococo del grupo B como presentación de sepsis neonatal tardía. An Pediatr (Barc). 2004; 60: 75-79.
- Nso Roca AP, Baquero-Artigao F, García-Miguel MJ, Del Castillo Martín F. Parotiditis aguda neonatal por Streptooccus agalatiae. An Pediatr (Barc). 2007; 67: 65-67.
- Brook I. Suppurative parotitis causedby anaerobic bacteria in newborns. Pediatr Infect Dis J. 2002; 21: 81-82.
Síndrome de shock tóxico estafilocócico como manifestación inicial de una osteomielitis
Sr. Director:
El síndrome de shock tóxico estafilocócico (SSTE) se define como una enfermedad aguda y grave, secundaria a la acción de toxinas producidas por algunas cepas de Staphylococcus aureus1-3. Se presenta un caso de SSTE de evolución atípica como manifestación inicial de una osteomielitis.
Niña de 8 años de edad, que acude a urgencias por presentar fiebre de 38 ºC, de 12 horas de evolución, asociada a escalofríos, cefalea, dolor abdominal y vómitos. En la exploración física presenta un regular estado general, palidez mucocutánea, taquicardia leve con presión arterial normal, signos meníngeos positivos e inyección conjuntival bilateral no supurativa, así como una herida en proceso de cicatrización en el segundo dedo del pie derecho. En el resto de la exploración no se detectan alteraciones.
En las pruebas complementarias destacan los siguientes parámetros: 10.900 leucocitos (99,5% neutrófilos), proteína C reactiva (PCR) 21 mg/L y procalcitonina normal (<0,2 ng/mL). La punción lumbar resultó normal. Se decidió el ingreso de la paciente para su observación con tratamiento sintomático. Durante las primeras 24 horas persistieron el regular estado general, la fiebre y la taquicardia. A las 36 horas desarrolló un exantema eritematoso de inicio en el tronco, que se generalizó hasta convertirse en eritrodermia. En la analítica de control destacaban los siguientes resultados: 18.500 leucocitos (91,9% neutrófilos), PCR 221 mg/L, GOT 98 UI/L, GPT 125 UI/L y bilirrubina total 2,6 mg/dL. Doce horas después presentó una hipotensión arterial, con valores inferiores al percentil 5 para la edad y la talla. Ante la sospecha de SSTE, se inició antibioterapia empírica intravenosa con cloxacilina 100 mg/kg/día y clindamicina 30 mg/kg/día. En el hemocultivo crecieron cocos Gram positivo en racimo. Tras 24 horas de tratamiento presentó una mejoría significativa del estado general, con desaparición de la fiebre y normalización de las constantes hemodinámicas. Al quinto día de ingreso inició una descamación en los pulpejos de las manos y los pies, y presentó dolor en el tobillo izquierdo con impotencia funcional, no referido hasta entonces. Se realizó una ecografía articular, en la que destacaba una pequeña colección en la articulación proximal del cuarto metatarsiano.
Se realizó una gammagrafía ósea, que confirmó la presencia de una osteomielitis en el tobillo izquierdo (figura 1). En el hemocultivo y el exudado nasal se aislaron colonias de S. aureus sensible a meticilina. La determinación de toxina del síndrome de shock tóxico (TSST-1) fue positiva. El estudio del gen de la leucocidina de Panton-Valentine (LPV) fue negativo. La evolución del caso clínico fue satisfactoria tras completar 6 semanas de antibioterapia oral con cefadroxilo.
El SSTE es una enfermedad muy rara en pediatría (con una incidencia de 0,5 casos por 100.000 habitantes)1-3. Se suele presentar de forma aguda y se considera la forma más grave de infección por S. aureus1. A pesar de ello, la mortalidad en niños es baja (un 3-7% de los casos)1-3. La sinusitis, la neumonía y las infecciones cutáneas o de herida quirúrgica constituyen el origen más frecuente1-3. Aunque cualquier foco de infección por S. aureus puede ocasionar este síndrome, son muy pocos los casos descritos en la bibliografía de SSTE asociado a osteomielitis4-6.
Las cepas de S. aureus causantes del SSTE son productoras de toxinas, fundamentalmente de enterotoxinas A, B y C y TSST-11-3,7,8. La LPV suele ocasionar infecciones más agresivas, con una gran destrucción y necrosis tisular. Su producción se asocia a S. aureus resistente a meticilina adquirido en la comunidad, aunque también se ha descrito en S. aureus sensible a meticilina5,9.
Ninguna prueba de laboratorio confirma la enfermedad. Su diagnóstico se basa en los criterios clínicos y microbiológicos propuestos por los Centers for Disease Control and Prevention3. Esta paciente cumplía dichos criterios, por lo que se consideró un caso confirmado de SSTE. Sin embargo, llama la atención su evolución atípica, lenta y poco virulenta, que no precisó medidas de soporte vital en una unidad pediátrica de cuidados intensivos. Diferentes estudios sugieren que la TSST-1 se asocia a casos menos agresivos y con una mortalidad inferior que la LPV5,9. Hasta un 20% de individuos sanos están colonizados por S. aureus, lo que podría originar en ellos la presencia mantenida de anticuerpos frente a la TSST-110. Este hecho podría explicar la escasa agresividad de la enfermedad en esta paciente. Algunas publicaciones recientes mencionan la posibilidad de que aparezcan casos de SSTE como el aquí descrito, menos virulentos, que podrían pasar desapercibidos o confundirse con otras entidades. Es probable que la incidencia real del SSTE sea mayor que la descrita3.
El mejor conocimiento de la enfermedad en los últimos años ha supuesto una anticipación en el diagnóstico y el tratamiento, lo que podría explicar la disminución de su morbimortalidad3,4. A pesar de su escasa frecuencia, en cualquier niño con fiebre, eritrodermia e hipotensión debe plantearse el diagnóstico diferencial con el SSTE e iniciar el tratamiento de forma precoz1-3.
Bibliografía
- Chuang YY, Huang YC, Lin TY. Toxic shock syndrome in children: epidemiology, pathogenesis and management. Paediatr Drug. 2005; 7: 11-25.
- Nso Roca AP, Menéndez Suso JJ, Riesco Riesco S, Benito Gutié¬rrez M, Ruza Tarrío FJ. Toxic shock as an initial sign of acute lymphoblastic leukaemia. An Pediatr (Barc). 2008; 69: 589-591.
- Costa Orvay JA, Caritg Bosch J, Morillo Palomo A, Noguera Julián T, Esteban Torne E, Palomeque Rico A. Toxic shock syndrome: experience in a pediatric intensive care unit. An Pediatr (Barc). 2007; 66: 566-572.
- Rey M, Wölfel D, Scharf J, Zeilinger G, Plettl-Maar J. Toxic shock syndrome due to osteomyelitis. Klin Padiatr. 1991; 203: 178-183.
- Dohin B, Gillet Y, Kohler R, Lina G, Vandenesch F, Vanhems P, et al. Pediatric bone and joint infections caused by Panton-Valentine leukocidin-positive Staphylococcus aureus. Pediatr Infect Dis J. 2007; 26: 1.042-1.048.
- Jacobson RM, Baltimore R. Toxic shock syndrome associated with osteomyelitis in premenarcheal girl. Pediatr Infect Dis J. 1989; 8: 125-126.
- John CC, Niermann M, Sharon B, Peterson ML, Kranz DM, Schlie¬vert PM. Staphylococcal toxic shock syndrome erythroderma is associated with superantigenicity and hypersensitivity. Clin Infect Dis. 2009; 49: 1.893-1.896.
- Ferry T, Thomas D, Bouchut JC, Gerard L, Vasselon-Raina M, Dauwalder O, et al. Early diagnosis of staphylococcal toxic shock syndrome by detection of the TSST-1 Vbeta signature in peripheral blood of a 12-year-old boy. Pediatr Infect Dis J. 2008; 27: 274-277.
- Lo WT, Lin WJ, Tseng MH, et al. Risk factors and molecular analysis of Panton-Valentine leukocidin-positive methicillin-resistant Staphylococcus aureus colonization in healthy children. Pediatr Infect Dis J. 2008; 27: 713-718.
- Liu GY. Molecular pathogenesis of Staphylococcus aureus infection. Pediatr Res. 2009; 65: 71R-77R.
Neumonitis intersticial crónica del lactante
Sr. Director:
Las enfermedades pulmonares intersticiales (EPI) en la infancia constituyen un grupo heterogéneo de enfermedades con muy escasa prevalencia (3,6 casos por millón de niños)1, por lo que inicialmente fueron clasificadas según los conocimientos extraídos de pacientes adultos. Posteriormente se puso de manifiesto que existen formas de EPI propias de la infancia y que el curso de las descritas en el adulto también es diferente en el niño, donde la respuesta al tratamiento es mayor y con mejor pronóstico. Estas peculiaridades podrían deberse a las diferentes etapas de desarrollo y maduración de los pulmones2. Entre los años 1997 y 2002, un grupo de neumólogos pediátricos de la Sociedad Respiratoria Europea (ERS)3 reevaluaron los expedientes de 185 niños con EPI, clasificándolos en cuatro grupos principales de diagnóstico: a) enfermedad parenquimatosa pulmonar de causa conocida (p. ej., neumonitis por hipersensibilidad); b) neumonía intersticial idiopática (p. ej., neumonía intersticial descamativa, neumonía intersticial linfocítica, neumonía intersticial no específica); c) otras formas de neumonía intersticial (p. ej., sarcoidosis), y d) trastornos congénitos (p. ej., disfunción del surfactante y linfangiectasia). Posteriormente se formó un grupo multidisciplinario, denominado Children's Interstitial Lung Disease (chILD), que estableció los siguientes criterios diagnósticos de EPI4: 1) presencia de síntomas respiratorios (tos, respiración difícil, intolerancia al ejercicio); 2) signos (taquipnea, crepitantes, retracciones, acropaquias, estancamiento ponderal, insuficiencia respiratoria); 3) hipoxemia, y 4) alteración difusa en la imagen radiológica. En 2002, la American Thoracic Society (ATS) y la ERS propusieron una clasificación de consenso, describiendo como formas clínicas específicas de la infancia la taquipnea persistente con hiperplasia de células endocrinas, la glucogenosis intersticial pulmonar, la enfermedad intersticial causada por alteraciones genéticas de las proteínas del surfactante y la neumonitis intersticial crónica del lactante.

Presentamos el caso de un varón de mes y medio de vida, sin consanguinidad ni otros antecedentes familiares de interés, y con líquido amniótico meconial en el parto como único dato obstétrico reseñable, hospitalizado tras el nacimiento por un cuadro de insuficiencia respiratoria compatible con taquipnea transitoria del recién nacido. A los 35 días de vida reingresó por presentar desnutrición y dificultad respiratoria de pocas horas de evolución, sin fiebre asociada. En la exploración física destacaba un aspecto desnutrido, un tiraje subcostal y una hipoventilación generalizada. Tras los primeros estudios complementarios realizados se descartó la existencia de fibrosis quística, déficit de alfa-1-antitripisina, inmunodeficiencia y neumonía por aspiración. Se realizó una fibroscopia flexible para descartar malformaciones en las vías respiratorias superiores, así como un tránsito esofagogastroduodenal, que resultaron normales. Se solicitó la realización de un proteinograma y un estudio de inmunoglobulinas y poblaciones linfocitaras, con resultados dentro de la normalidad, y una medición de la concentración intracelular de adenosín trifosfato, que demostró una respuesta celular inmunitaria moderada. En la reacción en cadena de la polimerasa no se detectó virus de la inmunodeficiencia humana, y el resto de serologías virales en sangre fueron irrelevantes. La radiografía de tórax mostró un patrón intersticial bilateral (figura 1) y en la tomografía computarizada torácica (figura 2) destacaban infiltrados en vidrio esmerilado bibasales. Ante la sospecha de EPI, se realizó una biopsia pulmonar, que mostraba cambios de fibrosis septal, hiperplasia de neumocitos, zonas de descamación y algún histiocito, datos compatibles con una neumonitis intersticial crónica del lactante. Se recogieron cultivos a partir de las muestras, que fueron negativos para Pneumocystis jiroveci, Criptococcus y micobacterias; en general, no creció ningún microorganismo. A partir de estos resultados, se instauró tratamiento con corticoides (orales y bolos) e hidroxicloroquina. Pese a ello, el paciente sufrió un empeoramiento progresivo, con aumento de la dificultad respiratoria y de las necesidades de oxígeno. Secundariamente, presentó un síndrome de Cushing y dificultad para la alimentación, por lo que fue necesario realizar una gastrostomía percutánea. Se planteó la posibilidad de realizar un trasplante pulmonar, por lo que se contactó con un centro de referencia, pero finalmente el paciente falleció a los 7 meses de vida en el contexto de una sobreinfección por Pseudomonas aeruginosa. Los resultados de la necropsia confirmaron la existencia de una neumonitis intersiticial crónica del lactante. No se hallaron alteraciones en otros órganos.

La neumonitis intersticial crónica del lactante es una de las formas de EPI específicas de la infancia, descrita por primera vez por Katzenstein et al.5 en 1995, a partir de los hallazgos en 9 pacientes no clasificables en ninguna de las formas descritas hasta el momento. El inicio de los síntomas se sitúa entre los 15 días y los 11 meses de vida, y los más frecuentes son la tos, la taquipnea y el estancamiento ponderoestatural. La clínica y la existencia de un patrón radiológico compatible son altamente sugestivos de EPI, pero el diagnóstico definitivo es anatomopatológico. En la biopsia se muestra que las alteraciones más significativas son el engrosamiento de los septos alveolares, la infiltración por células mesenquimales, la hiperplasia de neumocitos tipo 2 y el incremento de macrófagos intraalveolares6. La etiología se desconoce, pero podría ser secundaria a una respuesta alterada a la infección en un pulmón inmaduro7. Los corticoides son el pilar fundamental del tratamiento médico8; puede emplearse prednisolona o prednisona en una dosis de 1-2 mg/kg/día durante 6-12 semanas, o metilprednisolona en bolos de 10-30 mg/kg/día i.v. durante 3 días consecutivos mensualmente. Pueden asociarse otros fármacos, aunque los más empleados son la cloroquina y la hidroxicloroquina. La tasa de respuesta al tratamiento de la neumonitis intersticial crónica del lactante es inferior que la de la EPI en niños, valorada como entidad general (40-54%)9. El trasplante pulmonar sigue siendo el tratamiento definitivo y la única esperanza de supervivencia para estos pacientes, pero muchos fallecen antes de que sea posible realizarlo.
Bibliografía
- Dinwiddie R, Sharief N, Crawford O. Idiopathic interstitial pneumonitis in children: a national survey in the United Kingdom and Ireland. Pediatr Pulmonol. 2002; 34(1): 23-29.
- Clement A, Eber E. Interstitial lung diseases in infants and children. Eur Respir J. 2008; 31(3): 658-666.
- Clement A; ERS Task Force. Task force on chronic interstitial lung disease in immunocompetent children. Eur Respir J. 2004; 24(4): 686-697.
- Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, Bean JA, et al. Diffuse lung disease in young children. Application of a novel classification scheme. Am J Respir Crit Care Med. 2007; 176: 1.120-1.128.
- Katzenstein ALA, Gordon LP, Oliphant M, Swender PT. Chronic pneumonitis of infancy. A unique form of interstitial lung disease occurring in early childhood. Am J Surg Pathol. 1995; 19: 439-447.
- Vázquez Cordero C. Neumonitis intersticial crónica del lactante. An Esp Pediatr. 2002; 56 Supl 2: 54-58.
- Barbato A, Panizzolo C. Chronic interstitial lung disease in children. Paediatr Respir Rev. 2000; 1(2): 172-178.
- Kottmann RM, Hogan CM, Phipps RP, Simer PJ. Determinants of initiation and progression of idiopathic pulmonary fibrosis. Respirology. 2009; 14: 917-933.
- Dinwiddie R. Treatment of interstitial lung disease in children. Paediatr Respir Rev. 2004; 5: 108-115.
Hamartoma fibrolipomatoso congénito
El hamartoma fibrolipomatoso congénito precalcáneo es una entidad de la infancia de naturaleza benigna, que aparece en el nacimiento o unos meses después, en forma de una lesión nodular, unilateral o, más frecuentemente, bilateral y simétrica, en la parte posteromedial de los talones. Normalmente son lesiones asintomáticas y no se asocian otras alteraciones. Habitualmente no es necesario realizar ningún tratamiento.
Morbilidad de los niños prematuros en edad escolar (I): alteraciones neurosensoriales, psicointelectivas y de conducta
Objetivos: Conocer las alteraciones motoras, neurosensoriales, psicointelectivas, emocionales y de conducta en niños muy prematuros y prematuros tardíos en edad escolar.
Pacientes y métodos: Estudio de cohortes históricas de niños prematuros nacidos en el Hospital Clínico de Valladolid desde enero de 1996 hasta diciembre de 2001. Se incluyeron todos los recién nacidos (RN) con un peso al nacimiento ≤1.500 g y una edad gestacional (EG) ≤32 semanas (RN muy prematuros [RNMP]), y un grupo de niños con una EG de 33-36 semanas (RN prematuros tardíos [RNPT]). Se incluyó también una cohorte de RN a término (RNT) en el mismo periodo. Se citó a los niños en la consulta para realizar una entrevista sobre los problemas de salud y una exploración física completa a cada niño. Los datos de rendimiento escolar se tomaron de la entrevista a los padres. La valoración psicointelectiva y conductual fue realizada por una psicóloga infantil.
Resultados: Participaron en el estudio 35 RNMP, 44 RNPT y 40 RNT. La incidencia de parálisis cerebral fue del 11,4% en el grupo de RNMP. Tres de los RNMP tenían secuelas neurosensoriales graves: dos hipoacusia y uno ceguera bilateral. El coeficiente intelectual (CI) de los RNMP fue significativamente más bajo que el de los RNPT (91,23 ± 18,5 frente a 104,94 ± 16,2; p= 0,002) y que el de los RNT (91,23 ± 18,5 frente a 107,08 ± 15,4; p= 0,000). En el 15,4% de los RNMP el CI fue muy bajo (<69), y ninguno de los niños de los otros dos grupos se encontraba en este nivel. Se observó una correlación significativa entre el CI y el perímetro cefálico en el momento del alta hospitalaria en los RNMP. Respecto a la psicomotricidad (valorada mediante el test de Ozerestky), presentaron una peor coordinación general los RNMP en relación con los otros dos grupos. Los problemas de conducta fueron más evidentes en los niños prematuros tardíos. El 37,1% de los RNMP presentaban un mal rendimiento escolar y precisaban clases de apoyo, frente al 18,2% de los RNPT y el 7,5% de los RNT (p= 0,005). El 42,9% de los RNMP mostraban secuelas globales: un 11,4% graves, un 17,1% moderadas y un 14,3% leves. En el grupo de RNPT la prevalencia de secuelas fue del 27,3%, y en el de RNT del 20% (p <0,001); en ambos grupos eran leves.
Conclusiones: En la edad escolar, aproximadamente la mitad de los RNMP presentan alteraciones motoras, neurosensoriales, cognitivas o de conducta respecto a un grupo de RNT. Los prematuros tardíos presentaron una mayor incidencia de trastornos emocionales y/o de conducta que los RNMP y los RNT.
Faringoamigdalitis aguda por Mycoplasma pneumoniae. ¿Etiología infrecuente o infradiagnosticada?
Sr. Director:
La faringoamigdalitis aguda (FAA) es una de las enfermedades más frecuentes en la infancia, y es motivo de numerosas consultas y gran parte de las prescripciones antibióticas en niños1. El cuadro clínico característico asociado a Mycoplasma pneumoniae es la neumonía atípica, aunque también puede producir infecciones asintomáticas, procesos respiratorios altos y manifestaciones extrarrespiratorias2. La frecuencia y la importancia de este agente como responsable de la FAA han sido muy discutidas. Estudios basados en la determinación de la reacción en cadena de la polimerasa en tiempo real (PCR-RT) le otorgan un papel emergente3, mientras que estudios previos, basados exclusivamente en serologías o cultivos, consideran M. pneumoniae como una causa excepcional de FAA4,5.
Presentamos el caso de un niño de 2 años de edad, sin antecedentes personales de interés, que consulta por la presencia de fiebre de hasta 40,2 ºC, de 5 días de evolución, asociada a odinofagia e hiporexia, sin tos ni rinorrea y sin respuesta al tratamiento con penicilina V. En la exploración presentaba adenopatías submandibulares bilaterales >1 cm, amígdalas hipertróficas e hiperémicas con exudado blanquecino bilateral, e irritabilidad sin signos meníngeos. No se detectaron exantemas ni hepatoesplenomegalia. Presentaba una leucocitosis de 16.300 leucocitos (70,6% neutrófilos) y una proteína C reactiva (PCR) >250 mg/L. El test rápido de estreptococo, adenovirus e Influenza en exudado faríngeo resultó negativo. La radiografía de tórax era normal, así como el análisis sistemático de orina. Se realizó un hemocultivo, un urocultivo y serologías (CMV, EBV, Toxoplasma, VHH6, VHH7, parvovirus B19). Ante los elevados valores de la PCR, la persistencia de la clínica y la ausencia de mejoría con penicilina V, se decidió el ingreso del paciente y administrar tratamiento intravenoso con amoxicilina-ácido clavulánico.
Durante la primera semana de ingreso la exploración no se modificó, persistían picos febriles bifásicos, las analíticas no presentaban cambios y la PCR permanecía persistentemente elevada (>250 mg/L). Los cultivos resultaron estériles, las serologías negativas, la ecografía abdominal normal, y la ecografía de cuello era compatible con una adenitis inespecífica. El resultado de la lectura de Mantoux fue igual a 0 mm. Ante la persistencia de una PCR elevada y dado que en las últimas semanas se habían diagnosticado varios casos de neumonía por M. pneumoniae en nuestro servicio, se inició antibioterapia empírica con azitromicina. La fiebre desapareció a las 24 horas, y la exploración y la analítica se normalizaron. La serología mediante enzimoinmunoanálisis al noveno día de ingreso fue positiva para M. pneumoniae (títulos de IgM >1/64). Un mes después se confirmó la infección, con títulos IgM >1/256. Existen pocos casos publicados de infecciones por M. pneumoniae sin afectación de las vías respiratorias bajas o sintomatología catarral6. La PCR-RT permite realizar un diagnóstico rápido, aunque requiere equipo y personal especializados y es una técnica cara7. Un estudio publicado en 2007 analizó 140 niños diagnosticados de neumonía atípica, de los cuales un 32% presentó PCR-RT positiva, valor que ascendió hasta el 87% en los casos en que la duración de la enfermedad fue mayor (2-7 días).
Los autores concluyen que la PCR-RT podría ayudar a establecer un diagnóstico precoz de las infecciones por M. pneumoniae8.
Esposito et al.9 refieren una prevalencia de FAA por M. pneumoniae del 23,9%, una vez descartada la etiología estreptocócica, y empleando para ello tanto la serología como la PCR-RT. Un estudio reciente concluye que M. pneumoniae debe tenerse en cuenta sobre todo si existe clínica de broncoespasmo2. Algunos estudios previos han mostrado resultados discrepantes. Reed et al.4 encuentran prevalencias muy bajas (2%) en series en que el diagnóstico se realiza exclusivamente mediante serología y/o cultivo, mientras que en el trabajo de McMillan et al.5 los porcentajes fueron superiores (15,8%), pero sin diferencias significativas frente a un grupo control de niños sin FAA, por lo que concluyen que el aislamiento de M. pneumoniae no tiene significado patológico. Sin embargo, otras publicaciones recientes que utilizan conjuntamente PCR-RT y serología sí demuestran la capacidad patógena de M. pneumoniae y encuentran diferencias significativas frente a niños sin FAA2,10.
Clásicamente, M. pneumoniae se ha considerado una causa infrecuente de FAA, o incluso como un colonizador sin relevancia patológica. Sin embargo, las nuevas pruebas diagnósticas demuestran un papel importante de dicho germen. Aunque el fármaco de primera elección en la FAA bacteriana es la penicilina V, es frecuente la prescripción incorrecta de azitromicina1. Se presenta un caso que cursó con fiebre prolongada y que precisó hospitalización y la realización de diversas pruebas complementarias. Es infrecuente el estudio serológico de M. pneumoniae en las FAA con test de estreptococo negativo, y aun realizándolo, la IgM suele ser negativa hasta cumplir la primera semana del cuadro clínico. Por otro lado, ¿qué porcentaje de las FAA tratadas de forma incorrecta con azitromicina y con una evolución favorable podrían corresponder a M. pneumoniae, sobre todo teniendo en cuenta los elevados porcentajes de resistencia frente al estreptococo betahemolítico del grupo A? Creemos probable que no se trate de un caso excepcional, sino infradiagnosticado por las limitaciones de las técnicas microbiológicas disponibles actualmente. Otra limitación es que este microorganismo puede aislarse en la garganta como un patógeno primario, como copatógeno asociado a virus respiratorios o en portadores asintomáticos. La evolución benigna de las infecciones por M. pneumoniae no parece justificar el uso sistemático de técnicas caras, como la PCR-RT. Sin embargo, en casos con una mala evolución clínica, como el aquí presentado, tanto la serología como la PCR-RT pueden ayudar a establecer el diagnóstico.
Creemos que sería interesante realizar más estudios e implementar nuevas técnicas para intentar aclarar el verdadero papel de M. pneumoniae en la etiología de la FAA.
1. Ochoa C, Vilela M, Cueto M, Eiros JM, Inglada L; Grupo Español de Estudio de los Tratamientos Antibióticos. Adecuación del tratamiento de la faringoamigdalitis aguda a la evidencia científica. An Pediatr (Barc). 2003; 59: 31-40.
2. Fernández de Sevilla M, Alayeto J, Fernández Y, Muñoz-Almagro C, Luaces C, García-García JJ. Baja prevalencia de la infección por Mycoplasma pneumoniae en niños con faringoamigdalitis aguda. Enferm Infecc Microbiol Clin. 2009; 27: 403-405.
3. Principi N, Esposito S. Emerging role of Mycoplasma pneumoniae and Chlamydia pneumoniae in paediatric respiratory tract infections. Lancet Infect Dis. 2001; 1: 334-344.
4. Reed BD, Huck W, Lutz LJ, Zazove P. Prevalence of Chlamydia trachomatis and Mycoplasma pneumoniae in children with and without pharyngitis. J Fam Pract. 1988; 26: 387-392.
5. McMillan JA, Sandstrom C, Weiner LB, Forbes BA, Woods M, Howard T, et al. Viral and bacterial organisms associated with acute pharyngitis in a school-aged population. J Pediatr. 1986; 109: 747-752.
6. Dionisio D, Valassina M, Uberti M, Fabbri C, Parri F, Saffi EG. Mycoplasma pneumoniae non-pulmonary infection presenting with pharyngitis, polyarthritis and localized exanthem. Scand J Infect Dis. 2001; 33: 782-783.
7. Álvez F, Sánchez JM. Faringoamigdalitis aguda. Protocolos de Infectología 2009 de la Asociación Española de Pediatría [consultado en noviembre de 2010]. Disponible en: http://www.aeped.es/ documentos/protocolos-infectologia
8. Liu FC, Chen PY, Huang F, Tsai CR, Lee CY, Wang LC. Rapid diagnosis of Mycoplasma pneumoniae infection in children by polymerase chain reaction. J Microbiol Immunol Infect. 2007; 40: 507-512.
9. Esposito S, Cavagna R, Bosis R, Droghetti R, Faelli N, Principi N. Emerging role of Mycoplasma pneumoniae in children with acute pharyngitis. Eur J Clin Microbiol Infect Dis. 2002; 21: 607-610.
10. Esposito S, Blasi F, Bosis S, Droghetti R, Faelli N, Lastrico A, et al. Aetiology of acute pharyngitis: the role of atypical bacteria. J Med Microbiol. 2004; 53: 645-651.
Ectasia ductal mamaria. A propósito de dos casos
Sr. Director:
La ectasia ductal mamaria es una manifestación poco común en los niños, que en ocasiones conlleva la aparición de una telorragia importante, cuya presencia puede causar una gran alarma entre los progenitores. No obstante, en la edad pediátrica un alto porcentaje de las patologías que conllevan telorragia tienen un pronóstico excelente y son de carácter autolimitado.
Presentamos dos casos de ectasia ductal mamaria.
Caso 1
Lactante de 8 meses de edad, que acude al servicio de urgencias por presentar una ingurgitación mamaria y un sangrado intermitente por el pezón derecho de una semana de evolución. No tenía antecedentes familiares, obstétricos ni neonatales de interés. No recibía lactancia materna desde el segundo mes de vida. Los padres no referían antecedentes traumáticos ni de manipulación en la niña, y negaban la ingesta de fármacos.
En la exploración la niña mostraba un excelente estado general y estaba afebril. En la inspección no presentaba anomalías en el área mamilar, pero se observaba una induración en la palpación de la areola derecha, que emitía una sustancia serosanguinolenta. Dicha zona no presentaba signos de sobreinfección ni dolor con la manipulación. El resto de la exploración por aparatos y sistemas fue normal.
Ante la sospecha clínica de una ectasia ductal, se solicitó una ecografía mamaria, en la que no se observaban lesiones nodulares ni otras alteraciones; tampoco se apreciaban asimetrías ecográficas entre las dos mamas. El cultivo de la secreción no aisló gérmenes sugerentes de sobreinfección.
Se derivó a la paciente al servicio de endocrinología pediátrica para efectuarle un seguimiento, y en pocos meses se observó una resolución espontánea de la afección.
Caso 2
Niña de 13 meses de edad, que acude al servicio de urgencias por presentar una ingurgitación mamaria con emisión de sangre de forma intermitente por ambos pezones de dos semanas de evolución.
No tenía antecedentes familiares, obstétricos y neonatales de interés. No recibía lactancia materna desde los 4 meses de vida. Los padres no referían antecedentes traumáticos ni de manipulación en la niña, y negaban la ingesta de fármacos.
En la exploración estaba afebril. No se apreciaban alteraciones ni asimetrías en las mamas, los pezones y las areolas. Se observaba una secreción espontánea bilateral de una sustancia sanguinolenta. La palpación no provocaba dolor en las areolas, y no se observaban signos de rubor ni de calor local. El resto de la exploración por aparatos y sistemas fue normal.
Ante la sospecha clínica de una ectasia ductal, se solicitó una ecografía, en la que se observaban unas glándulas mamarias de 1 cm de tamaño aproximadamente, con formaciones quísticas anecoicas, más acusadas en el lado derecho. Al igual que en el caso anterior, no se aislaron gérmenes patógenos en el cultivo de la secreción.
Tras confirmar el diagnóstico se realizó un seguimiento clínico y ecográfico, y el cuadro se normalizó al cabo de 4 meses.
Aunque la galactorrea es un fenómeno que aparece con frecuencia en los neonatos y los lactantes en los primeros meses de vida, la telorragia en los lactantes o en niños más mayores es una patología rara que en ocasiones produce una gran alarma entre los familiares y el personal sanitario.
La causa más común de telorragia en estos pacientes es la ectasia ductal mamaria1-3.
La fisiopatología de esta entidad aún no está clara. Al parecer, el paso de hormonas maternas podría favorecer su desarrollo. Así, los estrógenos promoverían el crecimiento del sistema ductal, y la progesterona el de los alvéolos mamarios. La hormona foliculoestimulante y la prolactina también parecen tener un papel activo, ya que se han encontrado concentraciones particularmente altas de estas sustancias en el plasma de estos pacientes, que permanecen inalteradas en los primeros años de vida4,5.
Así pues, algunas series, como la de McKierman et al., han demostrado que gracias a estas hormonas las glándulas mamarias permanecen activas durante los primeros años de vida6.
Respecto al manejo terapéutico en los niños asintomáticos, se recomienda realizar un tratamiento conservador con controles ecográficos periódicos7,8. Se aconseja efectuar un cultivo de la secreción y, si existen signos sugerentes de sobreinfección, comenzar con antibioterapia empírica, teniendo en cuenta que el germen más frecuentemente implicado en dicho proceso es Staphylococcus aureus9. Se desaconseja la manipulación o el masaje del área afectada, ya que tales maniobras conllevan una ulceración del epitelio ductal y el consiguiente sangrado, y favorecen la colonización bacteriana.
A diferencia del manejo de la telorragia en los adultos, en los niños se debe evitar la realización de biopsias u otras pruebas invasivas para descartar un proceso maligno, siempre que no existan otros signos sugestivos aparte del sangrado3,7.
1. Miller JD, Brownell MD, Shaw A. Bilateral breast masses and bloody nipple discharge in a 4-year-old boy. J Pediatr. 1990; 116: 744-747.
2. Weimann E. Clinical management of nipple discharge in neonates and children. J Paediatr Child Health. 2002; 39: 155-156.
3. Bober E, Ozer E, Akgur F, Buyukgebiz A. Bilateral breast masses and bloody nipple discharge in a two year old boy. J Pediatr Endocrinol Metab. 1996; 9: 419-421.
4. Fenster DL. Bloody nipple discharge. J Pediatr. 1984; 104: 640-641.
5. Sigalas J. Bloody nipple discharge in infants. J Pediatr. 1985; 107: 484.
6. McKiernan JF, Hull D. Prolactin, maternal oestrogens, and breast development in the newborn. Arch Dis Child. 1981; 56: 770-774.
7. Stringel G, Perelman A, Jiménez C. Infantile mammary ductal ectasia: a cause of bloody nipple discharge. J Pediatr Surg. 1986; 21: 671-674.
8. Menken KU, Roll C. Bloody nipple discharge in a three year-old girl. Eur J Pediatr. 1992; 152: 1.047.
9. Imamoglu M, Ali C. Bloody nipple discharge in children: possible etiologies and selection of appropriate therapy. Pediatr Surg Int. 2006; 22: 158-163.
Exantema periflexural asimétrico
El exantema periflexural asimétrico en la infancia es un exantema de causa desconocida, caracterizado por una erupción maculopapulosa escarlatiniforme, con un crecimiento centrífugo. Comienza típicamente cerca de la axila o la región inguinal, de forma predominantemente unilateral, aunque a medida que la erupción progresa suele extenderse de forma bilateral. Se resuelve de manera espontánea en 1-3 semanas. Presentamos el caso de un niño de 4 meses de edad con esta erupción.









