
Hepatoblastoma de presentación neonatal
El hepatoblastoma es el tumor hepático maligno que aparece con más frecuencia en niños menores de 5 años, con predominancia por el sexo masculino. Su tasa de supervivencia a 5 años es del 70%, con resultados comparables en neonatos. Se ha descrito su asociación con determinados síndromes y, clínicamente, son más a menudo asintomáticos. Los marcadores tumorales alfafetoproteína y gonadotropina coriónica humana beta son muy útiles para el diagnóstico y seguimiento tras el tratamiento, si bien no son patognomónicos de este tipo de tumores. Para el diagnóstico se requiere la realización de pruebas de imágenes y una biopsia confirmatoria. El tratamiento se basa en la combinación de cirugía y quimioterapia, reservando la radioterapia para casos de mal pronóstico.
Calcificaciones cerebrales en el diagnóstico precoz del síndrome de DiGeorge sin cardiopatía
Tortícolis congénita como forma de presentación del síndrome de Klippel-Feil
Manifestaciones pulmonares en la neurofibromatosis tipo 1: una asociación a tener en cuenta
La neurofibromatosis tipo 1 es la enfermedad neurocutánea más frecuente. Es un trastorno genético con herencia autosómica dominante que produce alteraciones principalmente en la piel y en el sistema nervioso, pero también en otros órganos. La afec-tación pulmonar en pacientes con neurofibromatosis se ha descrito como una complicación rara que aparece principalmente en adultos. Presentamos el caso de un adolescente no fumador con neurofibromatosis tipo 1 y manifestaciones pulmonares asocia-das.
Los defectos en las extremidades, una facies peculiar y el fallo de medro no siempre constituyen un síndrome de Cornelia de Lange
Las malformaciones de las extremidades son raras y su etiología, variable. Existen síndromes genéticos que combinan estas malformaciones con discapacidad intelectual y otras malformaciones graves, como el síndrome de Cornelia de Lange. Presentamos el caso de una paciente con malformaciones severas en las 4 extremidades, discapacidad intelectual y características faciales peculiares que llevaron al diagnóstico presuntivo de síndrome de Cornelia de Lange. Presentamos el caso de una niña de 4 años de edad, padres consanguíneos y etnia gitana, con antecedentes de retraso del crecimiento intrauterino, bajo peso al nacer, microcefalia y múltiples malformaciones en ambas manos y pies, incluida la mano derecha hendida, con diagnóstico presuntivo de síndrome de Cornelia de Lange. Durante el primer año de vida se realizaron varios estudios con los siguientes resultados: cariotipo 46, XX; estudio de deleciones subteloméricas (técnica MLPA) normal; ecocardiograma y electrocardiograma sin hallazgos; evaluación oftalmológica y auditiva normales; ultrasonido abdominal y transfontanelar normales. A los 4 años se le aplicó una técnica de array de hibridación genómica comparada (comparative genomic hybridization) (array-CGH) con resolución de 180 kb, que detectó una deleción causal de 8,4 Mb en la citobanda 2q31.1q31.2. La deleción de 2q31 está asociada a la malformación de mano/pie hendido, y la correlación genotipo-fenotipo indica que las deleciones intersticiales de la región 2q31.1 muestran malformaciones en los miembros si incluyen una región crítica de 2,5 Mb, que incluye el cluster de HOXD y las regiones adyacentes en sentido 5´y 3´. Concluimos que ante un paciente con malformaciones graves y signos y síntomas superpuestos de varios síndromes, es aconsejable comenzar el plan de trabajo con array-CGH, y si esta técnica no está disponible, realizar un cariotipo de alta resolución con la intención de descartar reordenamientos cromosómicos.
Importancia de los tumores lipomatosos benignos en el niño como marcadores de anomalías estructurales y síndromes complejos. Experiencia en nuestro centro
Comunicación científica (XXIV). Lenguaje médico (2): Los epónimos en el lenguaje médico de la pediatría
Los epónimos son muy frecuentes en ciencias de la salud y una parte fundamental del lenguaje y de la cultura histórica de los médicos, ya que numerosas personas han dado nombre a enfermedades, síndromes y signos, partes anatómicas y procesos fisiológicos y patológicos. A pesar de ser términos etimológicamente vacíos, están ampliamente arraigados en la educación médica y en la historia de la medicina, de manera que sería muy difícil prescindir de ellos, o incluso, como proponen algunos, erradicarlos. En la actualidad no es frecuente que surjan nuevos epónimos médicos, ya que todos los desarrollos importantes de la medicina son fruto del trabajo en equipo y, por ello, es difícil bautizarlos con nombres que incluyan a todas las personas involucradas. Continúa el debate científico en la literatura médica con argumentos a favor y en contra de su uso. Lo razonable parece ser mantener los epónimos clásicos que han perdurado en el tiempo, sea porque tienen mayor importancia clínica, sensibilidad, especificidad y significación diagnóstica, o por su mayor importancia histórica.
Estudio descriptivo de las neumonías recurrentes en una unidad de neumología infantil
Introducción: Las neumonías recurrentes (NR) se definen como la presencia de infiltrados radiológicos que recurren después de una resolución completa del proceso neumónico inicial. A pesar de que son un motivo frecuente de demanda de asistencia en las unidades de neumología infantil, existen pocos estudios que evalúen su incidencia real.
Pacientes y métodos: Estudio descriptivo de los pacientes diagnosticados de NR en la Unidad de Neumología Infantil del Hospital Regional Universitario «Carlos Haya» (Málaga), tanto ambulatoriamente como en planta de hospitalización, durante un periodo de 1 año. Se procedió a la elaboración de una base de datos a través de un cuestionario de evaluación.
Resultados: Se incluyó a 157 pacientes con diagnóstico de NR, el 61,8% de los cuales fueron atendidos en consultas externas y el 37,6% en planta de hospitalización. Se estableció un diagnóstico etiológico en el 97,5% de los casos, y los más frecuentes fueron el asma (37,2%), la fibrosis quística (23,7%) y el síndrome aspirativo (11,5%). En el 75,3% de las recurrencias la localización fue variable, en el 14,3% única y en el 9,7% tuvo una distribución intersticial.
Conclusiones: La historia clínica y la exploración física detalladas, junto con las características radiológicas, orientan el diagnóstico de las NR en la mayoría de los casos. Elegiremos eventuales pruebas complementarias en función de la localización de las recurrencias, la edad del paciente y la prevalencia de las distintas patologías. En nuestro estudio, de forma global, las causas subyacentes de NR más frecuentes fueron el asma, la fibrosis quística y los síndromes aspirativos crónicos.
Síndrome PFAPA: de la sospecha al diagnóstico
La fiebre es uno de los principales motivos de consulta en la infancia. Generalmente, se asocia a procesos infecciosos banales, pero cuando se prolonga en el tiempo o los episodios recurren, es necesario hacer el diagnóstico diferencial con infecciones poco habituales, enfermedades autoinmunes, inmunodeficiencias primarias y neoplasias. El síndrome PFAPA (fiebre periódica, faringitis, estomatitis aftosa y adenitis) pertenece al grupo de síndromes de fiebre periódica, y se caracteriza por episodios febriles que aparecen regularmente durante meses alternándose con intervalos asintomáticos. Es la causa más frecuente de fiebre periódica en la infancia, y actualmente es una entidad clínica bien definida. Los corticoides son el pilar fundamental del tratamiento; resuelven el episodio de manera llamativa, hecho que constituye un criterio diagnóstico. El diagnóstico precoz y el tratamiento adecuado permiten mejorar la calidad de vida del paciente y su entorno, evitando la toxicidad farmacológica. Presentamos los casos de 2 niñas en edad preescolar con síndrome PFAPA.
Neurotóxicos medioambientales (IV)*. Tabaco, alcohol, solventes, flúor y aditivos alimentarios: efectos adversos en el sistema nervioso fetal y posnatal y medidas preventivas
Objetivos: 1) Divulgar la neurotoxicidad del humo del tabaco, alcohol y otros solventes, flúor y algunos aditivos alimentarios, y 2) recomendar las medidas preventivas para minimizar/eliminar su exposición.
Materiales y métodos: Revisión bibliográfica sistemática de los efectos en el sistema nervioso central (SNC) en desarrollo. Búsqueda en MEDLINE, Science Citation Index y Embase de los trabajos observacionales de exposición a bajas dosis en humanos y de experimentación en animales de los últimos 10 años.
Resultados: 1) El tabaquismo activo y pasivo de las madres gestantes provoca trastornos del aprendizaje, déficit de atención y del cociente intelectual (CI) persistente, y está asociado con un menor rendimiento académico en la descendencia; 2) la exposición fetal a bajas dosis de alcohol se ha asociado con hiperactividad, trastornos de atención, de aprendizaje y deterioro de la memoria en la descendencia; 3) la exposición a solventes por hobbies o aficiones en el hogar puede ser un factor de riesgo considerable, especialmente en áreas mal ventiladas; 4) estudios en animales y humanos sugieren que la exposición al flúor, a los niveles a que se expone la población por fluoración del agua potable y otros suplementos, puede tener efectos adversos sobre el neurodesarrollo, y 5) en animales de experimentación los efectos neurotóxicos por aspartamo y glutamato requieren dosis mucho mayores que las de la dieta humana.
Conclusiones: 1) El SNC fetal e infantil es especialmente vulnerable a la exposición a bajas dosis de humo de tabaco y alcohol; 2) no existe un nivel seguro de exposición ni para el tabaco ni para el alcohol; 3) el registro en la consulta de los hobbies o aficiones en el hogar con solventes permitirá detectar familias en riesgo; 4) los suplementos de flúor sólo están indicados en poblaciones de riesgo; 5) la relación entre la dieta y el comportamiento en niños con trastornos de déficit de atención e hiperactividad es incierta, y 6) la historia ambiental pediátrica es necesaria para avanzar en el conocimiento y en los aspectos preventivos, pronósticos y evolutivos de las enfermedades relacionadas con estas exposiciones.
Síndrome de Van der Woude
El síndrome de Van der Woude se caracteriza por la presencia de hoyuelos (pits) o fístulas labiales, asociados a hendiduras labiales y/o palatinas. También se han asociado otras anomalías congénitas, como defectos cardiacos y anomalías en los miembros. Es un trastorno genético con herencia autosómica dominante, con penetrancia casi completa y expresividad variable, estando implicados dos locus génicos. En este artículo repasamos la prevalencia, la etiología y los aspectos clínicos característicos de este síndrome, además de presentar el caso de un recién nacido varón con labio leporino bilateral y apéndices mucosos en el labio inferior. Insistimos en la importancia de la asociación de las hendiduras faciales junto a los pits, por el patrón hereditario del síndrome, así como el diagnóstico diferencial de distintos cuadros clínicos que presentan hoyuelos mucosos.
Lesiones faciales acneiformes en niña de 5 años
La esclerosis tuberosa es una enfermedad neurocutánea de herencia autosómica dominante y expresividad variable que afecta a la diferenciación y proliferación celular con la formación de hamartomas en distintos órganos. A pesar de que se han identificado dos genes, TSC1 y TSC2, situados en los cromosomas 9:9q34 y 16:16p13, respectivamente, el diagnóstico de la enfermedad es clínico ya que los tests genéticos tienen un porcentaje elevado de falsos negativos (15%). Las manifestaciones clínicas varían enormemente, incluso entre familiares. La presencia de lesiones cutáneas características es de gran utilidad para orientar el diagnóstico. El enfoque debe ser multidisciplinario ya que puede existir afectación renal, pulmonar, cardiaca o del sistema nervioso central con importante morbilidad. Es importante prestar atención a dichas complicaciones sistémicas para minimizar las secuelas de la esclerosis tuberosa.
Trago accesorio
El trago accesorio se trata de un nódulo del color de la piel, sésil o pediculado, único o múltiple, unilateral o bilateral. Su localización clásica es la región preauricular, pero puede localizarse en la región mandibular o cervical. Se debe establecer el diagnóstico diferencial, fundamentalmente con quistes epidermoides, fístulas y fibromas. En ocasiones, aparece asociado a otras anomalías del desarrollo de los arcos branquiales, como el síndrome de Goldenhar. El tratamiento quirúrgico responde a razones estéticas y conlleva la escisión completa del cartílago asociado.
Pilomatricomas múltiples. Implicaciones diagnósticas y terapéuticas
El pilomatricoma es el segundo tumor benigno superficial más frecuentemente extirpado en la infancia1. Su aspecto clínico característico es casi diagnóstico. La presencia de múltiples pilomatricomas nos debe hacer descartar ciertas patologías asociadas a este tumor. Presentamos dos casos de pilomatricomatosis múltiple.
«Situs inversus totalis» y cardiopatía congénita: asociación infrecuente que cabe tener en cuenta
Las anomalías en la disposición espacial de las vísceras toracoabdominales son entidades de muy baja prevalencia. Los síndromes heterotáxicos o situs ambiguos se asocian muy frecuentemente con cardiopatías congénitas graves, mientras que en el caso del situs inversus totalis (imagen especular con respecto a la normalidad) éstas se observan en el 3-5% de los pacientes, unas 6 veces más que en la población general. Se expone el caso de una niña de 26 meses de edad, portadora de dos defectos septales interventriculares en presencia de situs inversus totalis, y se incide en la importancia de realizar un estudio cardiológico ante anomalías de situs, así como en la necesidad de tener en cuenta la posibilidad de asociación de discinesia ciliar primaria con el trastorno visceral que presentamos.
Bebé colodión: manejo y proceso diagnóstico
El «bebé colodión» es una situación clínica poco frecuente que se presenta en el neonato y que es compartida por varias enfermedades y síndromes.
La alteración de la barrera epidérmica hace que el manejo y soporte del neonato sean fundamentales en los primeros días de vida, para luego llegar a un diagnóstico adecuado.
Presentamos un nuevo caso de esta entidad en la que la colaboración del neonatólogo y el dermatólogo es fundamental.
Síndromes de deficiencia de adhesión leucocitaria
Los síndromes de deficiencia de adhesión leucocitaria (leukocyte adhesion deficiency [LAD]) engloban un conjunto de patologías causadas por defectos en el reconocimiento, la adhesión y la migración de los leucocitos mieloides hacia los lugares de invasión microbiana, lo que provoca la falta de defensa innata del huésped frente a bacterias, hongos u otros microrganismos. Se distinguen dos tipos: LAD I y LAD II. Los síndromes LAD I y sus variantes están causados por mutaciones que impiden la expresión o la función de las integrinas de la clase -2 (integrinas CD11/CD18, o integrinas leucocitarias). Por el contrario, los sujetos con LAD II tienen unas características clínicas similares, pero conservan intacta la expresión y la función de las integrinas leucocitarias. El fundamento molecular de la deficiencia de tipo LAD II es un defecto en la glucosilación de los ligandos situados en los leucocitos, reconocidos por la familia de las moléculas de adhesión de las selectinas. Recientemente, se ha atribuido el defecto a mutaciones en un transportador de fucosa localizado en el aparato de Golgi. El establecimiento de las bases moleculares de los síndromes LAD ha generado nuevas perspectivas en el estudio de los mecanismos de acumulación leucocitaria, que son de gran relevancia en múltiples síndromes de inmunodeficiencias, así como en ciertas enfermedades inflamatorias que acaban desencadenando una lesión tisular.
Recién nacida con nevo melanocítico congénito gigante «en calzón»
El nevo melanocítico congénito gigante es una lesión pigmentada de gran tamaño presente al nacimiento. Su incidencia es de 1/1.000-500.000 recién nacidos. La localización más frecuente es el tronco posterior, la cara, el cuero cabelludo y las extremidades. Puede tener una morfología curiosa «en bañador» o «capelina».
Se presenta el caso de una recién nacida con un nevo congénito gigante «en calzón», que ocupaba toda la zona genital, inguinal, glútea, superior de ambos muslos e inferior del tórax, con pigmentación color marrón oscuro y negro, e hipertricosis, así como 3 más pequeños en las zonas parietal izquierda, antebrazo derecho y mentón. Además, presentaba lesiones planas pigmentadas en las extremidades y el tórax. Las analíticas, las ecografías abdominal y cerebral, la radiografía de raquis óseo y la resonancia magnética medular eran normales. El resultado de la biopsia de piel afectada fue de nevo intradérmico.
La mayor complicación del nevo melanocítico congénito gigante, aparte del problema estético, es la malignización. Estos nevos pueden asociarse con melanosis neurocutánea hasta en un 25% de los casos. Es fundamental el inicio del tratamiento lo más precoz posible.
Asociación de enfermedad celiaca y pancolitis ulcerosa: a propósito de un caso y revisión de la bibliografía
Presentamos el caso de una niña de 7 años y 6 meses de edad, diagnosticada de una enfermedad celiaca por presentar clínica, analítica y biopsia yeyunal compatibles, que mejoró tras instaurar una dieta exenta de gluten. Un año después presentó un cuadro de deposiciones con sangre, con analítica y estudio de colonoscopia sugestivo de pancolitis ulcerosa. Dado que la asociación de celiaquía y enfermedad inflamatoria intestinal apenas ha sido descrita en niños, nos parece interesante presentar el caso y llevar a cabo una revisión de la bibliografía disponible.
Mucopolisacaridosis tipo I: evolución clínica de 2 pacientes tras 30 meses de tratamiento enzimático sustitutivo
La mucopolisacaridosis tipo I (MPS I) es una enfermedad lisosomal hereditaria producida por un déficit enzimático de a-L-iduronidasa, que da lugar a una acumulación de los glucosaminglucanos (GAG) dermatán y heparán sulfato en los órganos y los tejidos, así como a un aumento de su excreción urinaria. Hay tres subtipos: la enfermedad de Hurler (MPS IH) es la más grave, la enfermedad de Scheie (MPS IS) es la más leve y la enfermedad de Hurler-Scheie (MPS IHS) es la forma intermedia.
Es una enfermedad crónica y progresiva, con manifestaciones multisistémicas. La disminución de la capacidad pulmonar y los síntomas de obstrucción de las vías altas (síndrome apnea-hipopnea del sueño), la afectación cardiovascular y los problemas articulares son los que causan mayor morbimortalidad.
El trasplante de médula ósea o de células madre hematopoyéticas, junto con el tratamiento enzimático sustitutivo, constituyen los principales pilares del tratamiento.
Presentamos 2 casos de MPS I que han recibido tratamiento sustitutivo con enzima recombinante a-L-iduronidasa durante 30 meses y un tercer caso de un paciente que lo ha iniciado hace 3 meses. Ninguno ha presentado complicaciones atribuibles al tratamiento.
Criptorquidia y otras anomalías del descenso testicular
La criptorquidia o testículo no descendido es el trastorno de las glándulas endocrinas masculinas más común en los niños. Se define como la falta de testículo en el escroto, secundaria a una anomalía en el proceso de descenso, y se engloba en el síndrome de escroto vacío. Su incidencia varía entre el 3,4 y el 5,8% en los niños nacidos a término.
Las razones más importantes para su tratamiento son: fecundidad disminuida, aumento de la tasa de neoplasias malignas, riesgo aumentado de torsión testicular o de lesión contra el pubis y estigma psicológico del escroto vacío.
La criptorquidia es una manifestación bien conocida de anomalías cromosómicas y un componente común de más de 50 síndromes de anomalías congénitas múltiples.
El diagnóstico se realiza por la anamnesis y el examen físico debiendo determinar situación, tamaño, comparación con el otro lado, desarrollo del escroto, movilidad, presencia o no de reflejo cremastérico, tamaño y morfología del escroto y del pene y signos de hernia inguinal.
Puede clasificarse por su ubicación en teste no palpable o palpable, que, a su vez, se diferencian en criptorquidia o ectopia testicular.
En los casos de testes no descendidos palpables no es necesaria una evaluación analítica para contribuir al diagnóstico.
La hormonoterapia se realiza con gonadotropina coriónica (hCG) y está recomendada en testículos inguinales distales o en aquellos que se encuentren en la entrada del escroto.
En el caso de testes palpables uni- o bilateralmente se realiza una orquidopexia; en el caso de los no palpables se recomienda una incisión inguinal inicial para la exploración adecuada del canal inguinal: si se aprecia un testículo de características normales, se realiza orquidopexia; si se localizan testes atróficos o restos testiculares, se realiza exéresis; si no se localizan restos en el canal inguinal, se realiza una inspección intrabdominal con la óptica de laparoscopia. Actualmente, se recomienda para el tratamiento del testículo intrabdominal la orquidopexia asistida por laparoscopia.
Tumor de Wilms en paciente trasplantado renal
Los pacientes portadores de trasplantes tienen un riesgo superior al resto de la población de padecer neoplasias. En el caso de los trasplantados renales éste es aún mayor, pues, además de tomar una medicación inmunodepresora, en muchas ocasiones han estado expuestos durante largos periodos de tiempo a técnicas de diálisis, con el consiguiente riesgo de haber desarrollado enfermedad renal quística adquirida. Se expone el caso de una niña de 6 años portadora de un trasplante renal normofuncionante desde los 2 años, que acude a urgencias por presentar un cuadro de dolor abdominal, diagnosticada finalmente de un tumor de Wilms en el riñón nativo izquierdo. Se incide sobre la utilidad de la ecografía en este tipo de pacientes (hay protocolos que recomiendan su realización periódica como método de cribado de procesos neoplásicos) y sobre la importancia de reconocer determinados síndromes o condiciones predisponentes al desarrollo de tumores en este grupo de pacientes, que modificarán la actitud terapéutica.
Síndrome de Rud: observación de un nuevo caso
La asociación de la tríada oligofrenia, ictiosis congénita e hipogonadismo constituye el síndrome de Rud, entidad con muy pocos casos en la literatura médica mundial. Se han descrito en estos pacientes otras alteraciones menos frecuentes, como crisis convulsivas, epilepsia, talla baja, retinitis pigmentosa, polineuropatía hipertrófica o sordera neurosensorial. Forma parte de los denominados síndromes neurocutáneos queratósicos, junto con otros mejor definidos, como el síndrome de Sjögren-Larsson o la enfermedad de Refsum. Se expone el caso de un paciente de 13 años de edad, con criterios clínicos compatibles con el síndrome de Rud, que asocia una agenesia renal unilateral y un sarcoma de partes blandas.
El complejo de esclerosis tuberosa: importancia del examen de la piel en la epilepsia
El complejo de esclerosis tuberosa (CET) es uno de los trastornos neurocutáneos más frecuentes en la edad pediátrica. Nuestro objetivo es presentar el caso clínico de un adolescente con epilepsia parcial compleja y esclerosis tuberosa. El interés del caso radica en la inexistencia de manchas cutáneas hipocrómicas, la ausencia de diagnóstico previo de CET, a pesar de la avanzada edad del paciente, y la presencia de criterios suficientes para establecer el diagnóstico.
Importancia de la mancha mongólica: síndromes asociados y diagnóstico diferencial
Sr. Director:
La mancha mongólica (MM), también llamada melanocitosis dérmica congénita, suele aparecer en el nacimiento o durante las primeras semanas de vida. Aumenta en los 2 primeros años y después desaparece de modo gradual. A los 10 años la mayoría de estas manchas han remitido; si la mancha se mantiene en la edad adulta, se denomina MM persistente (3-4% de los orientales). Su frecuencia, similar en ambos sexos, varía entre los distintos grupos raciales. El término «mancha mongólica» se debe a su frecuencia elevada en las razas orientales, sobre todo en los mongoles, en quienes aparece en el 90% de los recién nacidos. Se observa en el 80% de los individuos de raza negra, en el 40% de los latinos y en menos del 10% de los caucasianos y judíos1-3.
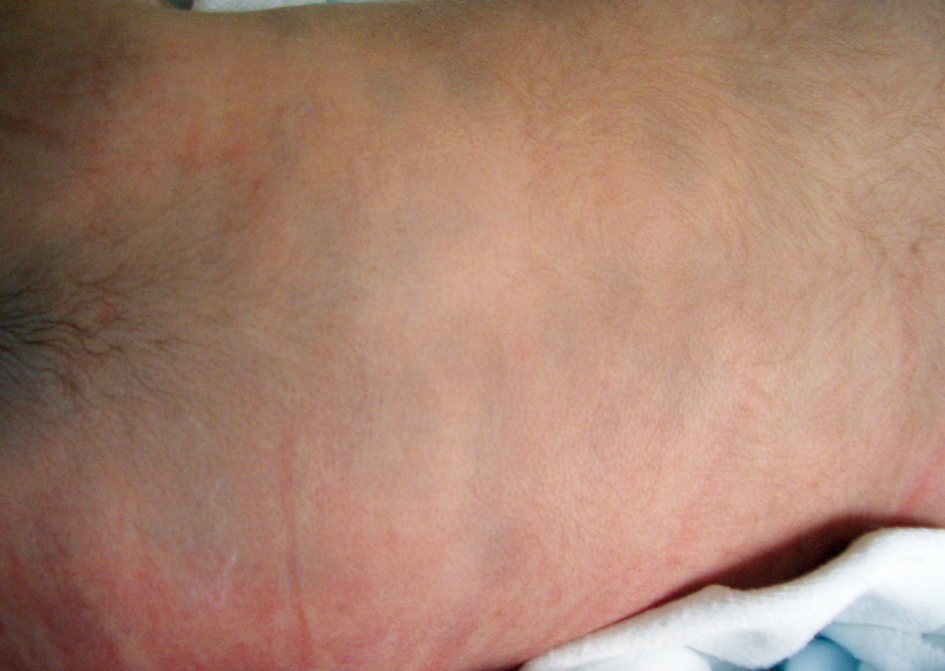
La localización clásica es la región lumbosacra y las nalgas. Se conoce como MM aberrante cuando se presenta en áreas atípicas, como la espalda, los hombros, el cuero cabelludo y las extremidades (figuras 1 y 2). Ésta es más probable que persista en la edad adulta. La forma extensa o generalizada no es excepcional, ya que aparece en más del 3% de los niños asiáticos, indios americanos y negros3-5.
Clínicamente, se presenta como una o varias máculas de morfología angulada, redonda u ovalada. El tamaño varía entre 1 y 20 cm y los bordes están mal definidos (las más grandes están mejor delimitadas). Tienen una coloración homogénea azul grisácea (figura 1) que no se acentúa en la exploración con lámpara de Wood. En personas de piel oscura adopta un tono verdoso (figura 2). La MM sobreimpuesta es una variante con zonas más oscuras en su seno3,6.
El estudio histológico muestra melanocitos dendríticos fusiformes dispersos entre los haces de colágeno de los tercios inferiores de la dermis. Esta melanocitosis dérmica se debe a que no se ha completado el proceso de migración de los melanocitos desde la cresta neural a la unión dermoepidérmica. Normalmente no hay melanocitos dérmicos desde la semana 20 de gestación. Las diferencias raciales podrían indicar la existencia de factores genéticos implicados en este proceso. La coloración azulada de las lesiones se debe al fenómeno de Tyndall: la pigmentación dérmica se ve azul, por la menor reflectancia de la región con longitudes de onda mayores en comparación con la zona que la rodea2,3.
No se han descrito casos de malignización de las MM. Dado que las máculas desaparecen con el paso de los años, no suelen necesitar tratamiento. En las lesiones persistentes se puede emplear maquillaje o laserterapia (con una mejor respuesta en MM aberrantes de pacientes de menos de 20 años)7.
La mayoría de las MM no tienen significación patológica. En ocasiones la presencia de formas aberrantes, extensas o múltiples puede facilitar el diagnóstico y posterior tratamiento precoz de ciertas enfermedades8:
1. Facomatosis pigmentovascular. Consiste en la combinación de una malformación vascular (casi siempre una mancha en vino de Oporto) con un lesión hiperpigmentada cutánea congénita (nevo epidérmico, nevo spilus o melanocitosis dérmica). Puede existir un nevo anémico. Se han clasificado en cinco tipos, que a su vez se han subdividido en a o b, en función de la ausencia o presencia de manifestaciones extracutáneas (como los síndromes de Sturge-Weber y de Klippel-Trenaunay). Aparece una MM en: a) facomatosis pigmentovascular tipo II (la más frecuente) asociada a una malformación capilar tipo mancha en vino de Oporto; b) tipo IV combinada con una mancha en vino de Oporto y un nevo spilus, y c) tipo V (facomatosis cesiomarmorata), que coexiste con cutis marmorata telangiectásica congénita4.
2. Gangliosidosis GM1, tipo 1 o infantil. Enfermedad debida a una deficiencia de la betagalactosidasa. En los primeros 6 meses de vida los pacientes presentan facies tosca, cuello corto, hipertrofia gingival, macroglosia, hipotonía, hipertricosis, retraso psicomotor y del desarrollo, convulsiones, hepatosplenomegalia, deformación de los cuerpos vertebrales, de la silla turca, anomalías de los huesos largos y de la pelvis, y manchas rojo cereza en la mácula. Los niños raramente sobreviven más de 3 años5,8.
3. Síndrome de Hurler. Mucopolisacaridosis tipo I, la más frecuente, producida por un déficit de alfa-L-iduronidasa. Comparte muchos rasgos con la gangliosidosis GM1 infantil. Alrededor de los 6 meses se observa opacidad corneal, visceromegalia, disostosis múltiple y rasgos faciales anormales. Provoca retraso mental, infecciones respiratorias y cardiopatía mortal alrededor de los 10 años de edad8.
4. Síndrome de Hunter. Mucopolisacaridosis tipo II, causada por un déficit de la enzima iduronato-2-sulfatasa y con un espectro de gravedad amplio. La forma grave cursa con síntomas similares al síndrome de Hurler9.
5. Labio leporino. Muchos niños japoneses con este defecto presentan una MM en la hendidura.
Se han descrito casos aislados en pacientes con enfermedad de Niemann-Pick, alfamanosidosis, síndrome de Sjögren-Larsson, hemangioma congénito, manchas café con leche (facomatosis pigmentopigmentaria), dedos supernumeraciones y melanosis leptomeníngea. Quizás algunas de estas asociaciones puedan ser casuales4,8.
El diagnóstico de la MM es fácil de establecer cuando la morfología clínica y la localización son típicas. En otras localizaciones podría confundirse con las siguientes entidades:
• Otras melanocitosis dérmicas, como el nevo azul (rara vez congénito y de localización muy variada), el nevo de Ota (sobre todo en la región periorbitaria, la sien, el área malar y la esclerótica ipsolateral), el nevo de Ito (regiones supraclavicular, escapular o deltoidea) y el hamartoma melanocítico dérmico (nalgas, manos y piernas)3.
• Otras neoplasias melanocíticas benignas, como la mancha café con leche, la melanosis de Becker y el nevo melanocítico congénito. El nevo melanocítico gigante, cuando afecta a la zona baja de la espalda y las nalgas, adopta un aspecto «en traje de baño». Difieren de la MM en el color y en los bordes bien definidos.
• Malformaciones vasculares, como las malformaciones glomovenosas o las manchas en vino de Oporto (se asocian a la MM en la facomatosis pigmentovascular) o los «precursores» de hemangiomas.
• Contusiones o hematomas. Éstos, si aparecen en lactantes de menos de 9 meses de edad y se sitúan en regiones típicamente protegidas, como las nalgas o la espalda, representan los signos físicos de maltrato infantil más frecuentes. Por tanto, una MM no diagnosticada o el desconocimiento de la variante aberrante, múltiple o sobreimpuesta podrían generar una sospecha errónea de maltrato. Cabe resaltar que el 40% de los neonatos de etnia gitana presentan MM (población con mayor incidencia de sufrir malos tratos según algunos estudios). Es curioso que el 12,5% de las madres atribuyan dichas manchas a un traumatismo durante el parto1. Por ello, es necesario anotar en la historia neonatal la presencia y la localización de las MM y proporcionar una correcta información a los padres10.
• Otras: argiria, ocronosis, eritema discrómico persistente (dermatosis cenicienta), hiperpigmentación postinflamatoria, hipermelanosis nevoide lineal y espiral, atrofodermia idiopática de Pasini y Pierini, y erupción fija medicamentosa.
En conclusión, la población española actual cuenta con más grupos raciales en que es frecuente la aparición de MM. Es importante conocer sus variantes (aberrante, persistente, múltiple, extensa o generalizada y sobreimpuesta), las enfermedades asociadas (facomatosis pigmentovascular, labio leporino y metabolopatías) y evitar su confusión con las lesiones de maltrato infantil.
Bibliografía
- Ikizoglu T, Ergör S, Asar GM, Yilmaz O. Frequency and characteristics of Mongolian spots among Turkish children in Aegean region. Turk J Pediatr. 2006; 48: 232-236.
- Avilés Izquierdo JA, Hernanz Hermosa JM, De la Cueva Dobao P. Mancha mongólica. Acta Pediatr Esp. 2004; 62: 60-61.
- Valdés F, Ginarte M, Toribio J. Melanocitosis dérmicas. Actas Dermosifiliogr. 2001; 92: 379-388.
- Torrelo A, Zambrano A, Happle R. Large aberrant Mongolian spots coexisting with cutis marmorata telangiectatica congenita (phacomatosis pigmentovascularis type V or phacomatosis cesiomarmorata). J Eur Acad Dermatol Venereol. 2006; 20: 308-310.
- Torrelo A, Solana LG, Mediero IG, Ruiz-Falco ML, García-Peñas JJ, Zambrano A. Mancha mongólica generalizada en un niño con gangliosidosis GM1 tipo 1. Actas Dermosifiliogr. 2000; 91: 31-33.
- Egemen A, Leung AK, Robson WL. Superimposed Mongolian spots. Pediatr Dermatol. 2008; 25: 233-235.
- Kagami S, Asahina A, Watanabe R, Mimura Y, Shirai A, Hattori N, et al. Laser treatment of 26 Japanese patients with Mongolian spots. Dermatol Surg. 2008; 34: 1.689-1.694.
- Ashrafi MR, Shabanian R, Mohammadi M, Kavusi S. Extensive Mongolian spots: a clinical sign merits special attention. Pediatr Neurol. 2006; 34: 143-145.
- Ochiai T, Suzuki Y, Kato T, Shichino H, Chin M, Mugishima H, et al. Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): a survey among 52 Japanese patients. J Eur Acad Dermatol Venereol. 2007; 21: 1.082-1.085.
- Al Jasser M, Al-Khenaizan S. Cutaneous mimickers of child abuse: a primer for pediatricians. Eur J Pediatr. 2008; 167: 1.221-1.230.
Síndromes polimalformativos con expresión en el sistema nervioso central
Objetivos: Determinar las características de los síndromes polimalformativos con expresión en el sistema nervioso central (SNC) detectados en el periodo neonatal, e investigar los factores de riesgo maternos asociados.
Material y métodos: Estudio retrospectivo, durante el periodo comprendido entre enero de 1976 y diciembre de 2005, realizado en el Servicio de Pediatría del Hospital Universitario de Guadalajara. Se incluyeron en el estudio todos los recién nacidos en nuestro hospital con síndromes polimalformativos con expresión en el SNC.
Resultados: Durante el periodo de estudio, se recogieron siete casos de niños con malformaciones congénitas del SNC y síndrome polimalformativo. Las anomalías más frecuentes fueron la hidrocefalia congénita y los defectos del tubo neural. Los factores de riesgo encontrados fueron las infecciones durante la gestación, la radiación ionizante, la toma de algunos fármacos y la consanguinidad.
Conclusiones: Los síndromes polimalformativos con expresión en el SNC tienen una etiología multifactorial, que comprende tanto factores ambientales como genéticos.
Síndromes de Pearson y de Kearns-Sayre: dos enfermedades mitocondriales multisistémicas, debidas a deleciones en el ADN mitocondrial
El síndrome de Pearson (SP) y el síndrome de Kearns-Sayre (SKS) son enfermedades mitocondriales multisistémicas con diferente fenotipo, causadas por deleciones en el ADN mitocondrial (ADNmt).
Objetivo: Describir las manifestaciones clínicas y los hallazgos neurorradiológicos, bioquímicos y genético-moleculares de ambos síndromes, con objeto de difundir su conocimiento entre los pediatras.
Pacientes y métodos: Se han estudiado retrospectivamente 6 pacientes con SKS y 3 con SP inicial, dos de los cuales evolucionaron a SKS.
Resultados: La edad de inicio de los síntomas fue inferior en los SP. Los síntomas más precoces fueron los hematológicos (anemia), seguidos de los renales (Fanconi) y digestivos (insuficiencia pancreática), y de forma más tardía se presenta la afectación ocular, endocrinológica, cardiológica y neurológica. Cuatro pacientes precisaron implantación de marcapasos. Seis casos presentaron alteraciones cerebrales y/o del tronco del encéfalo en la resonancia magnética. Se observó hiperlactatorraquia, hiperproteinorraquia y descenso de ácido fólico en el líquido cefalorraquídeo. La mitad de los SKS presentaron fibras musculares rojo-rasgadas y fibras citocromo C oxidasa negativas. En ocho pacientes se detectó una deleción única del ADNmt.
Conclusiones: 1) Las diferencias más acusadas entre el SP y el SKS fueron la edad de comienzo y las manifestaciones iniciales. Los síntomas en la evolución, así como los hallazgos bioquímicos, neurorradiológicos y genéticos, fueron similares. 2) Las enfermedades mitocondriales deberían incluirse en el diagnóstico diferencial del síndrome de Fanconi, el déficit de la hormona del crecimiento y los trastornos de la conducción cardiaca, especialmente en los casos con afectación multiorgánica. El diagnóstico se confirma por la presencia de una gran deleción en el ADNmt.
Herpes zóster con lesiones intensas y diseminación cutánea extensa en una niña inmunocompetente
El virus de la varicela zóster es el causante de dos síndromes diferentes: la varicela, como resultado de la infección primaria, y el herpes zóster (HZ), consecuencia de la reactivación del virus que permaneció latente en los ganglios sensitivos espinales.
Aunque el HZ se considera habitualmente una enfermedad rara y benigna durante la infancia, su incidencia aumenta en los niños inmunodeprimidos y en los que han padecido la varicela en el primer año de vida. Además, los niños con immunodeficiencia pueden presentar una afectación grave de los dermatomas infectados y HZ diseminado. Exponemos el caso clínico de una niña de 9 años de edad que presentó una erupción característica de HZ con lesiones intensas que afectaban a tres dermatomas contiguos y, además, una diseminación cutánea. Aunque la paciente no había padecido varicela, se registró en la familia la existencia de un ambiente epidemiológico de varicela durante el periodo neonatal.








