
Eficacia de la recomendación de un patrón de dieta mediterránea en preescolares con sobrepeso y obesidad
Introducción: Tras realizar algunas puntualizaciones −no suficientemente aclaradas en las guías de alimentación− hemos recurrido a la dieta mediterránea tradicional y hemos medido los valores antropométricos al comienzo y al finalizar el año del estudio en pacientes que hacían un cumplimiento razonablemente correcto de las guías, y aun así tenían sobrepeso y obesidad.
Pacientes y métodos: Se incluyeron niños de 2 a 8 años con sobrepeso y obesidad en un programa de educación nutricional denominado «Aprendiendo a comer del Mediterráneo». Para evaluar la calidad de la dieta, hemos utilizado el test Kidmed y para poder medir las nuevas propuestas hemos diseñado el test de la Dieta Mediterránea Tradicional.
Resultados: Se obtuvo una muestra de 92 pacientes (42 niñas y 50 niños). La edad media fue de 5,3 años. Tras un año de intervención dejaron de ser obesos el 75,7% y dejaron de tener sobrepeso el 74,3%. En conjunto, el percentil del IMC descendió 12,1 puntos. Se consiguió una mejoría en la calidad de la alimentación reflejada en una mejor puntuación de ambos índices. El test Kidmed no resultó apropiado para el control del sobrepeso.
Conclusiones: La aplicación de un patrón de dieta mediterránea tradicional en el ámbito familiar ha resultado muy efectiva en el control de niños con sobrepeso u obesidad. Consideramos que el cumplimiento del test de la Dieta Mediterránea Tradicional junto al programa nutricional «Aprendiendo a comer del Mediterráneo» han sido claves en los resultados.
Incidencia de errores innatos del metabolismo y otros trastornos detectados en un programa de cribado metabólico neonatal ampliado de un grupo mexicano de hospitales
Introducción: La detección de errores innatos del metabolismo (EIM), endocrinopatías, hemoglobinopatías y otros trastornos por medio del cribado metabólico neonatal es una iniciativa de salud mundial que comenzó hasta el año 1973 en México. La incidencia nacional de este grupo de enfermedades es incierta debido a la falta de programas de cribado metabólico neonatal ampliado (CMNA), aunada a la carencia de publicaciones relacionadas. Para el presente manuscrito, la incidencia de EIM en el noreste de México se estima a partir de un programa de CMNA en hospitales privados del Grupo Christus Muguerza.
Material y métodos: El estudio fue retrospectivo e incluyó la revisión de resultados de 19.768 recién nacidos (RN), obtenidos de marzo de 2006 a febrero de 2017.
Resultados: El programa de CMNA detectó a 60 RN con algún EIM u otro trastorno y 104 fueron identificados como heterocigotos, presentando una incidencia de 30,4 y 52,7 por cada 10.000 RN, respectivamente. El diagnóstico más frecuente fue la deficiencia de glucosa-6-fosfato deshidrogenasa (G6PD); y en el caso de los heterocigotos, las hemoglobinopatías. La combinación de tecnologías en el cribado resultó en una especificidad del 99,95%, una sensibilidad cercana al 100% y un valor predictivo positivo del 86,96%.
Conclusiones: Los programas de CMNA ofrecen la posibilidad de detectar y confirmar un diagnóstico temprano para ofrecer un tratamiento específico, en combinación con un asesoramiento genético. Por otro lado, estos resultados proporcionan una estimación de la incidencia de los EIM, endocrinopatías, hemoglobinopatías y otros trastornos en un grupo de hospitales privados en México.
Influencia de los estilos parentales en el consumo adolescente de tabaco, alcohol y medicamentos sin prescripción
Introducción: La manera en la que los padres se relacionan con sus hijos puede actuar como factor de protección o de riesgo ante el consumo de determinadas sustancias por los adolescentes. Los objetivos de este trabajo fueron comprobar la relación entre los consumos de tabaco, alcohol y medicamentos sin prescripción, la posible influencia de los estilos de socialización parental en dichos consumos y las diferencias de sexo en los mismos.
Sujetos y método: Determinación de las asociaciones entre los consumos de tabaco, alcohol y medicamentos sin prescripción y las dimensiones de los diferentes estilos de socialización parental a través del coeficiente de correlación de Pearson a partir de los datos obtenidos de 665 adolescentes con una media de edad de 15,5 años (DT= 1,1) en centros educativos de la Comunidad de Madrid.
Resultados: Se encontraron correlaciones positivas entre los consumos de tabaco, alcohol y medicamentos sin prescripción. Se observaron correlaciones negativas entre las dimensiones Afecto-comunicación y Normas inductivas de ambos progenitores y dichos consumos, y positivas con la dimensión Crítica-rechazo. Se hallaron según el progenitor y la sustancia correlaciones positivas entre el consumo y las Normas indulgentes y Normas rígidas, así como diferencias por sexo.
Conclusiones: El afecto, la comunicación y el control conductual actúan como factores protectores ante el consumo adolescente de sustancias con impacto negativo en la salud, mientras que la crítica, el rechazo o la laxitud en el control se constituyen como factores de riesgo en dicho consumo
Vacunación frente a rotavirus en niños prematuros: una cuestión de tiempo
La infección por rotavirus continúa siendo un problema relevante de salud pública en los países desarrollados, y los niños prematuros constituyen la población más vulnerable tanto en términos de riesgo de infección como por la gravedad de la misma. En la actualidad existen dos vacunas frente a rotavirus disponibles en España: la vacuna pentavalente bovina-humana reordenada (RotaTeq®, MSD) y la monovalente humana atenuada (Rotarix®, Glaxo-SmithKline Biologicals). Los datos de los ensayos clínicos y estudios observacionales indican que la vacunación anti-rotavirus en niños prematuros es bien tolerada y segura, con un perfil similar en este sentido al observado en niños nacidos a término. Por otra parte, los resultados de los estudios clínicos y observacionales también apoyan una eficacia similar a la comunicada en niños a término. Más aún, al tratarse de un grupo de mayor riesgo de infecciones, es posible teóricamente que el beneficio obtenido sea incluso mayor que en niños a término. La transmisión horizontal del virus vacunal, aun siendo posible, no ha sido documentada en ensayos clínicos y los estudios observacionales existentes hasta ahora sugieren un riesgo bajo. En consonancia con diversas recomendaciones nacionales e internacionales, consideramos que esta población, siempre que su condición clínica lo permita, debe recibir la vacunación frente a rotavirus sin retrasos, incluyendo su vacunación mientras estén hospitalizados, si así fuese necesario.
Evolución en 12 años de los indicadores de lactancia materna y factores asociados en un Centro de Salud acreditado IHAN
Introducción: La Organización Mundial de la Salud (OMS) recomienda la lactancia materna exclusiva 6 meses y la mantenida hasta 2 años o más. Nuestro objetivo es estudiar la evolución de indicadores de lactancia (duración, porcentajes de inicio y mantenimiento hasta los 12 meses) en los últimos 12 años en nuestro centro, acreditado recientemente como Centro de Salud IHAN, y la asociación de estos indicadores con distintos factores: educación maternal, forma de nacimiento (parto vaginal o cesárea) y tipo de maternidad (pública o privada).
Material y métodos: Estudio descriptivo prospectivo de cohorte con 803 lactantes nacidos en dos periodos de tiempo separados 12 años: 2003-2004 y 2015-2016. Datos recogidos por entrevista personal en las revisiones del niño sano.
Resultados: La mediana de duración de la lactancia ha mejorado de forma significativa en el segundo periodo (10 meses) respecto al primero (5 meses; p <0,001); también los porcentajes de lactancia al nacimiento y hasta 12 meses (p <0,05). La asistencia a educación maternal ha sido el factor que se asocia de forma más significativa a estos indicadores en ambos periodos; también el parto vaginal (en el primer periodo) y la maternidad pública (en el segundo). Estos resultados se confirman en el análisis multivariante.
Conclusiones: En estos 12 años se ha producido una mejora significativa en los indicadores de lactancia de nuestro centro. Los factores favorables para iniciar y mantener la lactancia materna son asistir a los cursos de educación maternal (el más importante), nacer por parto vaginal y en maternidad pública.
Fracaso escolar: algunas consideraciones para los pediatras / Asociación entre el uso de dispositivos con pantalla y la integridad de la sustancia blanca en niños preescolares
School-aged children who are not progressing academically: considerations for paediatricians
Rey-Casserly C, McGuinn L, Lavin A; AAP Committee on psychosocial aspects of child and family health. Section on Developmental and Behavioural Pediatrics. Pediatrics. 2019; 144: e20192520.
«El fracaso escolar es un gran problema que debiera ser abordado desde todos los ámbitos», apuntaba el buen pediatra Venancio Martínez en un editorial de Pediatría Integral en septiembre de 2014, sobre una observación de las cifras de fracaso escolar en España que nos colocan en los puestos de cabeza de la Unión Europea. En Estados Unidos las cifras se sitúan en entorno de la media europea. La Academia Americana de Pediatría (AAP) ha considerado que el fracaso escolar es también responsabilidad del pediatra y ha publicado unas consideraciones para los pediatras que pasamos a resumir.
Se estima que un 13% de los niños escolarizados tiene algún tipo de problema de aprendizaje. De estos, un 35% tendría algún problema específico de aprendizaje (entendido como una alteración en uno o más de los procesos psicológicos básicos que participan en el entendimiento y en el uso del lenguaje, que se manifiestan como una incapacidad para escuchar, pensar, hablar, leer, escribir, deletrear o hacer cálculos matemáticos), un 20% alteraciones del lenguaje y un 13% otros problemas de salud que limitan la capacidad de estar alerta o la vitalidad, como las cardiopatías, el asma o la epilepsia, entre otros. Un 9% de esos niños estaban diagnosticados de trastorno del espectro autista, un 6% de discapacidad intelectual y otro 6% de retraso del desarrollo psicomotor. Algo menos del 2% de los niños presentaba múltiples discapacidades.
Aunque la mayor parte de la responsabilidad para la evaluación y el manejo de estos niños con problemas académicos recae tradicionalmente en el sistema escolar, los pediatras, como garantes del bienestar y de la salud de los niños, contribuyen de forma importante. La AAP, a través de la Sección de Neurodesarrollo y Conducta, señala en este documento de posicionamiento el papel que debe corresponder al pediatra, que incluye las siguientes funciones:
• Prevención. Incluye la contribución a proteger a estos niños frente al daño cerebral. Las vacunaciones, el seguimiento del crecimiento y del desarrollo, el cribado de la anemia, la promoción del uso de cascos en las actividades deportivas, las medidas de fijación para los vehículos, la prevención en el uso de tabaco, alcohol y otras sustancias, junto con la identificación del riesgo psicosocial, son algunas de las herramientas que emplean los pediatras en su práctica habitual.
• Reconocimiento temprano de los problemas de aprendizaje. Debe realizarse una historia detallada previa en el periodo perinatal, del desarrollo y el comportamiento y del patrón de sueño. La recogida precisa de una historia familiar y social (p. ej., algunas veces las historias de trauma personal y la ansiedad pueden interferir en el aprendizaje), así como de múltiples cambios de colegio. Debe completarse con una exploración física exhaustiva, poniendo especial atención a las alteraciones neurológicas (tono, coordinación).
• Diagnóstico de las afecciones médicas subyacentes. En algunos niños, la historia y los hallazgos físicos revelan la necesidad de realizar pruebas adicionales, como el electroencefalograma, las pruebas de neuroimagen y los test genéticos y/o metabólicos.
• Remisión para evaluaciones más específicas. Es frecuente requerir la valoración por parte de psicólogos, neuropsicólogos o logopedas que evalúen las habilidades de capacidad cognitiva, el funcionamiento adaptativo de los logros académicos, las habilidades de comunicación, el funcionamiento motor, el estado emocional y/o de comportamiento de estos niños. A pesar de estas derivaciones, el pediatra acompañará a la familia y le ayudará a tomar decisiones.
Lo que aporta este trabajo:
Lo que tradicionalmente se ha considerado un problema de las familias y las escuelas, el fracaso escolar, es también un aspecto en el que el pediatra se debe ver involucrado. Por una parte, porque existen motivos directamente médicos por los que los niños pueden fracasar en el colegio. Pero, por otra, el fracaso escolar, además de otras situaciones del entorno escolar, está muy directamente relacionado con la salud. Como bien comenta Venancio Martínez, «el estudio del desarrollo en la edad preescolar, precisamente cuando nuestro contacto con los padres es más frecuente, debería detectar a estos niños antes de que puedan tener dificultades en la escuela». Asimismo, deberían participar más activamente en el asesoramiento del diseño de los planes educativos. El reto es grande e implica una mejora en los programas de formación de los pediatras en el periodo de residencia, pero también en la formación continuada, en especial en el ámbito de la atención primaria, además de una mayor cercanía a la realidad de los centros educativos. Abordajes como el de la AAP, a través de su estrategia «Bright Futures: Guidelines for health supervision of infants, children and Adolescents», pueden servir de guía para ello.
• Tratamiento. Algunos niños necesitan tratamientos médicos concretos, y otros más ayuda por parte de logopedas, fisioterapeutas o terapeutas ocupacionales. El pediatra debe ser el que coordine estos tratamientos. La meta última del tratamiento es permitir al niño alcanzar su máximo potencial, lo cual requiere muchas veces ciertas adaptaciones y modificaciones en la educación (regulado por ley). La evidencia revela que hacer repetir de curso a un niño no es una estrategia efectiva para ayudarle a encontrar su máximo potencial.
• Monitorización. Se utilizan instrumentos y cuestionarios estructurados para medir en los niños el funcionamiento cognitivo y adaptativo, el habla y el lenguaje, el estado conductual y emocional, las habilidades motoras finas y la coordinación en las evaluaciones. Los logros académicos del niño se valoran usando medidas estandarizadas. Es importante establecer un diálogo continuo entre los pediatras y las escuelas.
Un papel clave para el pediatra que atiende a niños con alteraciones de salud mental o del neurodesarrollo es ayudar a los adultos implicados en su educación a percibir apropiadamente las fortalezas y los desafíos que en ella se plantean. Estos niños pueden ser percibidos como perezosos o intencionadamente oposicionistas, más que como niños con un déficit neurocognitivo que les imposibilita alcanzar un progreso académico típico.
M.J. Galiano Segovia
Pediatra. Centro de Salud María Montessori. Leganés (Madrid)
Associations between screen based media use and brain White matter integrity in preschool-aged children
Hutton JS, Dudley J, Horowitz-Kraus T, DeWitt T, Holland SK. JAMA Pediatr. [DOI: 10.1001/jamapediatrics,2019.386].
En este estudio, los investigadores se plantearon confirmar científicamente si existen riesgos neurobiológicos debido al uso de dispositivos electrónicos en niños preescolares. Analizaron la asociación entre el uso de pantallas a esa edad y los índices de integridad y mielinización de la sustancia blanca, especialmente de los tractos relacionados con el lenguaje, las funciones ejecutivas y el inicio de la lectura. Partieron de la hipótesis de que un mayor tiempo de uso de pantallas estaría asociado con una menor mielinización en esas áreas cerebrales y con peores puntuaciones en los test que evaluaban dichas capacidades.
La investigación fue coordinada por el Dr. Hutton, pediatra del Hospital Infantil de Cincinnati. Se llevó a cabo un estudio transversal entre agosto de 2017 y noviembre de 2018. Completó todas las pruebas un total de 47 niños sanos de 3-5 años de edad, nacidos con, al menos, 36 semanas de edad gestacional, y sin factores de riesgo ni alteraciones en su neurodesarrollo.
Los padres rellenaron una encuesta sobre el uso de dispositivos con pantallas (Screen Q survey), que consta de 15 ítems en los que se valora el acceso a pantallas, la frecuencia de uso y el contenido visualizado, solos o en compañía de los padres. Las puntuaciones van de 0 a 26, y las más altas reflejan un mayor uso de dispositivos electrónicos.
A los niños se les realizaron test para valorar el lenguaje, la comprensión lectora y el procesamiento fonológico (CTOPP-2), test de denominación rápida de objetos, test de vocabulario expresivo (EVT-2) y test de habilidades prelectoras Get Ready to Read (GRTR).
Se completó el trabajo realizando a los niños estudios de neuroimagen mediante resonancia magnética dinámica, una potente técnica para cuantificar la integridad de la sustancia blanca cerebral, y en concreto valorar la organización y la mielinización de los tractos. Se valoraron especialmente las áreas relacionadas con el lenguaje. El fascículo arqueado co¬necta las áreas cerebrales receptiva (Wernicke) y expresiva (Broca) del lenguaje, y se asocia con habilidades como la fonología y el vocabulario. El fascículo uncinado y el fascículo longitudinal inferior se relacionan con el procesamiento semántico, la integración emocional y la incorporación de imágenes visuales.
Aunque las redes sensoriales maduran relativamente pronto, las conexiones para habilidades intelectuales superiores, como el lenguaje, las funciones ejecutivas y la lectura, tienen
un desarrollo un poco más tardío, y dependen mucho de la es¬timulación tanto en el hogar como en otros ambientes.
Resultados y conclusiones del estudio
Al analizar estadísticamente los resultados obtenidos en el estudio, se observó una asociación entre un mayor uso de pantallas en los niños preescolares y una menor mielinización de las áreas y tractos de sustancia blanca cerebral relacionados con el lenguaje, la función ejecutiva y las habilidades para la lectura. Es decir, los niños con un mayor uso de pantallas tenían una menor «maduración» en las áreas cerebrales que intervienen tanto en el lenguaje como en la lectura, así como puntuaciones más bajas en los test del lenguaje y vocabulario.
En este estudio se pone de manifiesto que, al menos en algu¬nos aspectos, el uso de dispositivos con pantallas durante la temprana infancia se acompaña de riesgos neurobiológicos.
Lo que aporta este trabajo:
Los autores responden a la demanda de la sociedad de apoyar consejos saludables en datos objetivos y con rigor científico. Es la primera vez que se publica un estudio con pruebas de neuroimagen sobre la repercusión del uso de pantallas en niños preescolares. Aunque la muestra no es muy amplia, hay que valorar el enorme esfuerzo que supone llevar a cabo este tipo de estudios en niños tan pequeños. Es difícil que los preescolares colaboren en los test, y las pruebas de neuroimagen no siempre pueden realizarse correctamente a esta edad.
Los resultados de este artículo apoyan las recomendaciones que tanto desde la Asociación Española de Pediatría, la Academia Americana de Pediatría y la Organización Mundial de la Salud se están realizando sobre la importancia de limitar el uso de pantallas en niños pequeños.
Se necesitan más estudios de este tipo para conocer cómo influye el uso de pantallas según las diferentes edades de los niños y confirmar su alcance a largo plazo.
C. Esteve Cornejo
Pediatra. Clínica Universidad de Navarra. Madrid
Uso de internet en los pacientes que acuden a consultas externas de pediatría de un hospital general
Introducción: La relación médico-paciente asiste a un nuevo paradigma: la búsqueda de información médico-sanitaria por los pacientes, en ocasiones antes o después de acudir a consulta médica.
Material y métodos: Estudio transversal, observacional, no experimental. Encuesta en las consultas externas de un hospital general de área a los padres/madres que acudían por primera vez.
Resultados: Contestaron 220 progenitores. El 90% disponían de conexión a internet en su domicilio y el 98% en su móvil. Solo el 38% buscaron información antes de acudir y el 25% la buscarían al llegar a su domicilio. El 34% de los encuestados encontró información relacionada con su problema y al 26% les resultó adecuada. Al 38% les hubiera gustado que tras la visita su médico les hubiese recomendado alguna página web de calidad y a un 80% les parecía una buena idea que el servicio de pediatría contase con su propia página web.
Conclusión: Cada día es más frecuente que los pacientes busquen información médico-sanitaria en internet antes de acudir a consulta y por ello los profesionales sanitarios y los propios servicios deben estar preparados para ofrecer información de calidad por estos medios.
Impacto de la realización de cursos de reanimación cardiopulmonar avanzada pediátrica en la isla de Lanzarote
Objetivo: Conocer el nivel actual de conocimientos de reanimación cardiopulmonar (RCP) pediátrica del personal sanitario de Lanzarote y analizar la eficacia de los cursos de RCP avanzada pediátrica en la isla.
Método: Estudio analítico transversal de los cursos de RCP avanzada pediátrica y neonatal realizados en la isla desde 2016 hasta 2018. Se recogieron las puntuaciones en las evaluaciones teóricas al inicio y al final del curso así como en las prácticas y se contrastaron con variables sociodemográficas del alumnado. Se aplicó la prueba de Wilcoxon para contrastar las puntuaciones teóricas antes y después del curso y modelos de regresión lineal múltiple para estudiar la relación entre desempeño y distintas variables sociodemográficas.
Resultados: 77 alumnos realizaron los cursos con una puntuación mediana que aumentó significativamente de la evaluación inicial (14 puntos) a la final (18 puntos) sobre 20 (p < 0,001). En la evaluación práctica tanto de RCP básica como de avanzada y neonatal, las puntuaciones medianas de cada una de las maniobras superaron el valor 4, siendo el mínimo exigido 3 sobre 5. Los médicos y los profesionales con mayor puntuación teórica inicial ejecutaron mejor las maniobras de RCP básica y avanzada (p <0,05). Los profesionales más jóvenes realizaron mejor la RCP avanzada (p <0,05).
Conclusiones: Los cursos de RCP avanzada pediátrica y neonatal son métodos docentes eficaces a corto plazo para la formación teórico-práctica de los profesionales sanitarios. Se precisan futuros estudios que midan el efecto a medio y largo plazo de los mismos.
Grupos psicoeducativos para padres de niños con trastorno por déficit de atención e hiperactividad
En el tratamiento del niño con trastorno por déficit de atención e hiperactividad (TDAH) debemos incluir desde el principio a la familia, principalmente a los padres, ya que a través de ellos nuestras intervenciones podrán tener una mayor repercusión y beneficio para el menor.
Proponemos como pilar de cualquier intervención los grupos psicoeducativos de padres, en los que se les da la información y las herramientas necesarias para comenzar a entender el problema de su hijo, cómo ayudarlo y las distintas alternativas de tratamientos, y desde ahí mejorar la alianza con los terapeutas que van a intervenir.
Se establece en el centro de salud mental un programa psicoeducativo de 4 sesiones de 2 horas cada una para padres con hijos con TDAH, dirigido por la psicóloga y la psiquiatra del programa infantojuvenil. Al final de cada grupo se hace una evaluación anónima para tener un feedback sobre cómo los padres habían recibido la información transmitida con 4 preguntas, la valoración de las sesiones y la posibilidad de aportar sugerencias.
Se hace un análisis de los cuestionarios recogidos durante un curso escolar, obteniéndose una valoración muy positiva en todos los aspectos sugeridos, y con la posibilidad de ampliarlos o incluir más temas en nuevas sesiones.
Estos resultados nos animan a mejorar esta alternativa e incluirla como base en todos los tratamientos de los niños con TDAH, y se propone a los padres que ante este diagnóstico inicien este grupo y desde ahí se plantee cada caso de forma individualizada.
Educación afectivo-sexual en adolescentes, una tarea de todos
El preocupante incremento de las infecciones de transmisión sexual (ITS) entre adolescentes en los últimos años, revela la necesidad de una autocrítica social que promueva la sensibilización y el abordaje conjunto de instituciones políticas, docentes, profesionales sanitarios y padres. La figura del pediatra de Atención Primaria como agente educador en salud afectivo-sexual podría ser clave en nuestro medio.
Proyecto internacional INSPIRE: «¿Qué es normal en la leche humana» (II). Compuestos inmunológicos solubles
La protección inmunológica del recién nacido depende principalmente de factores inmunitarios maternos proporcionados a través de la leche. Sin embargo, muy pocos estudios han evaluado la variabilidad natural de los diferentes compuestos inmunitarios presentes en la leche humana de mujeres sanas pertenecientes a poblaciones heterogéneas. En este contexto, el objetivo de este trabajo fue la detección y cuantificación de una amplia gama de factores inmunitarios solubles, entre los que se incluyen factores de inmunidad innata (IL-1β, IL-6, IL-12, INFγ, TNFα) y adquirida (IL-2, IL-4, IL-10, IL-13, IL-17), quimioquinas (IL-8, Groα, MCP1, MIP1β), factores de crecimiento (IL-5, IL-7, EGF, G-CSF, GM-CSF, TGFβ2) e inmunoglobulinas (IgA, IgG, IgM), en la leche producida por mujeres sanas de diversas etnias procedentes de distintos entornos geográficos, dietéticos, socioeconómicos y ambientales. A partir de los resultados de este trabajo, pudimos determinar que un grupo de estos factores (IgA, IgG, IgM, EGF, TGFβ2, IL-7, IL-8, Groα y MIP1β) estaba presente en todas o en la mayoría de las muestras recogidas en todas las cohortes y, por tanto, podría considerarse como el núcleo común (core) de la leche humana en condiciones fisiológicas.
El tiempo frente a una pantalla se asocia con problemas de inatención en preescolares: resultados del estudio de la cohorte de recién nacidos CHILD / Uso de concentrados de cannabis en adolescentes
Screen-time is associated with inattention problems in pre-schoolers: results from
the CHILD birth cohort study
Tamana SK, Ezeugwu V, Chikuma J, Lefebvre DL, Azad MB, Moraes TJ, et al. PLoS One. 2019; 14(4): e0213995.
M.J. Galiano Segovia
Pediatra. Centro de Salud María Montessori. Leganés (Madrid)
Cannanbis concentrate use in adolescents
Meier MH, Docherty M, Leischow SJ, Grimm KJ, Pardini D. Pediatrics. 2019; 144(3): e20190338.
C. Esteve Cornejo
Pediatra. Clínica Universidad de Navarra. Madrid
Proyecto internacional INSPIRE: «¿Qué es normal en la leche humana?» (I). Oligosacáridos de la leche humana
La leche humana es un fluido complejo, compuesto por una amplia variedad de sustancias, entre las que se incluyen nutrientes y otros componentes bioactivos. Los datos disponibles en la actualidad sugieren que, en condiciones fisiológicas, el número y la concentración de estas sustancias pueden variar en función de muchos factores (base genética, etnia, localización geográfica, dieta, tiempo posparto...). En este contexto, el objetivo del proyecto internacional INSPIRE era conocer la variabilidad natural en la composición de la leche humana entre mujeres sanas que difieren en cuanto a su localización geográfica, etnia, dieta y situación socioeconómica, que se reflejan en la existencia de grandes diferencias en los factores ambientales, microbiológicos y socioculturales relacionados con la crianza y el entorno del recién nacido. En este artículo se muestra el diseño general del estudio y los resultados obtenidos con los oligosacáridos de la leche humana (HMO), uno de los componentes mayoritarios en este fluido biológico, con funciones biológicas muy relevantes para la salud infantil. Los resultados muestran un claro efecto de la cohorte (p <0,05) sobre las concentraciones de casi todos los HMO. Además, la edad materna, el tiempo posparto, el peso y el índice de masa corporal se correlacionaron con varios HMO. Por otra parte, se observaron diferencias en el perfil de HMO entre poblaciones étnicamente similares pero que viven en lugares diferentes, lo que sugiere que los factores medioambientales pueden desempeñar un papel en la regulación de la síntesis de los diferentes HMO.
Actividad física y marcadores de salud cardiovascular en niños pequeños / Asociación entre la ingesta de gluten en los primeros 5 años de vida y anticuerpos pos
Hay abundante evidencia científica en población adulta que demuestra que la actividad física previene y enlentece la progresión de la ECV.
Los niños en edad escolar que son más activos tienen mejores indicadores de salud cardiovascular, como, por ejemplo, menor presión arterial (PA) en reposo, mejor estado de forma cardiovascular e índices más favorables de resistencia arterial. Estos hallazgos han propiciado que la Asociación Americana del Corazón (AHA) recomiende la actividad física como parte de la estrategia primaria en la prevención de la ECV. La actividad física puede retardar el declive en la salud cardiovascular, que comienza ya desde el nacimiento. Sin embargo, hay poca evidencia para apoyar esta recomendación en la infancia temprana (≤8 años).
El propósito del estudio Health Outcomes and Physical Activity in Preschoolers (HOPP), un ensayo observacional prospectivo, era determinar el efecto de la actividad física sobre los indicadores de salud cardiovascular durante la infancia temprana. En concreto, se valoró el efecto de la actividad física total y de la actividad física modera-intensa sobre el estado cardiovascular, la elasticidad arterial y la PA durante un periodo de 3 años. La hipótesis de trabajo era que los niños pequeños que realizan más actividad física y de mayor intensidad tienen mejores indicadores de salud cardiovascular.
Cada año, durante 3 años, los niños incluidos en la cohorte de estudio, completaban una serie de valoraciones distribuidas en 2 visitas separadas por 19 ± 14 días. En la primera visita se realizaba una antropometría y una valoración del estado de forma. Al final de la visita, al niño se le proporcionaba un monitor de actividad física (acelerómetro) para llevar durante 1 semana en la cadera derecha. Después del periodo de actividad monitorizada, el niño iba a la segunda visita, en la que se valoraba la elasticidad arterial en reposo y la PA. Los datos se recogieron entre agosto de 2010 y septiembre de 2014. En la visita inicial se obtenía el consentimiento informado por escrito de los representantes legales de los niños.
Se excluyeron del estudio los niños con cualquier patología o retraso en el desarrollo cognitivo. Completaron la visita inicial 418 niños; 42 participantes se perdieron durante el seguimiento.
La actividad física se midió mediante acelerómetros. Para valorar una perspectiva integral de la salud cardiovascular, se tuvieron en cuenta 3 indicadores relativos: a) indicador de estado de forma cardiovascular mediante prueba de esfuerzo (carrera en cinta) con medida de la recuperación de la frecuencia cardiaca; b) la elasticidad arterial mediante la velocidad de onda de pulso y el índice B de elasticidad de la arteria carótida, y c) PA sistólica medida en posición sentada.
La mayor duración de carrera en la cinta en los niños se correlacionó con la actividad física total y la actividad física modera-intensa, así como en el tiempo de recuperación de la frecuencia cardiaca basal. También se encontró una mejoría de la elasticidad arterial en función del ejercicio, pero sólo en la velocidad de onda de pulso. La actividad física moderada-intensa se asoció con una menor tasa de cambio en la PA sistólica sólo en las niñas. Los efectos eran independientes de la edad, la estatura o el índice de masa corporal.
Los autores concluyen que los niños que realizan mayor actividad física en edades tempranas tienen mejores índices de salud cardiovascular. A mayor intensidad de la actividad, menor rigidez en la pared arterial. Además, sugieren que la causa de esa mejoría puede estar mediada por una mejora de la función del sistema nervioso autónomo, al igual que ocurre en el sujeto adulto. La actividad física moderada-intensa también proporciona más beneficios (medidos como menor rigidez arterial).
Lo que aporta este trabajo:
Este estudio aporta evidencia de que la actividad física es beneficiosa para la salud cardiovascular, tanto en niños como en niñas, independientemente de la edad, la estatura o el índice de masa corporal, y rellena un importante agujero en la literatura, demostrando que los efectos protectores de la actividad física sobre la salud cardiovascular empiezan pronto en la infancia. Aunque los autores no especifican el tipo de actividad física que realizaron los participantes en el estudio, como pediatras hemos de reivindicar el papel que tiene el juego al aire libre –y las actividades lúdicas que comporta–, no sólo para la salud cardiovascular de nuestros niños, sino también para su desarrollo madurativo y emocional. Saltar a la comba, jugar en el patio al balón, montar en bicicleta o jugar al «pilla-pilla» no deben quedarse en el recuerdo de lo que eran las ocupaciones del tiempo libre de la generación de sus padres, sino que continúan siendo parte importante en la promoción de la salud de los niños, en especial de los más pequeños.
Pediatra. Centro de Salud María Montessori. Leganés (Madrid)
Andrén Aronsson C, Lee HS, Hård Af Segerstad EM, Uusitalo U, Yang J, Koletzko S, et al; TEDDY Study Group. JAMA. 2019;
322(6): 514-523 [DOI: 10.1001/jama.2019.10329] [PubMed PMID: 31408136; PubMed Central PMCID: PMC6692672].
Este artículo es fruto del seguimiento de la cohorte de niños que participaron en el estudio TEDDY (The Environmental Determinants of Diabetes in the Young). El estudio TEDDY consiste en el seguimiento desde el nacimiento hasta los 15 años de una cohorte de niños con riesgo genético de padecer enfermedad celiaca y diabetes mellitus, procedentes de 6 centros situados en Finlandia, Alemania, Suecia y Estados Unidos, con el fin de determinar el riesgo de aparición a lo largo del seguimiento de diabetes mellitus o enfermedad celiaca, en función de variables relacionadas con la dieta. En este caso, se investigó el riesgo de aparición de anticuerpos de celiaquía y de enfermedad celiaca en los 5 primeros años, en función de la cantidad de gluten consumida.
Los niños se incluyeron en el estudio al nacimiento (periodo de reclutamiento entre 2004 y 2010), y la determinación de riesgo genético se realizó mediante el estudio de los genotipos de HLA.
Se incluyeron 8.676 recién nacidos portadores de algunos de los genotipos HLA asociados a la enfermedad celiaca y diabetes mellitus. Para la determinación de la autoinmunidad se midieron los anticuerpos antitransglutaminasa anualmente desde los 2 años.
La ingesta de gluten se estimó a partir de un registro de ingesta de 3 días, que se recogió a los 6, 9 y 12 meses, y cada 2 años hasta llegar a los 5.
El objetivo primario fue una autoinmunidad positiva, definida como aumento en los niveles de antitransglutaminasa en suero en 2 determinaciones consecutivas. El objetivo secundario fue la confirmación de enfermedad celiaca mediante biopsia intestinal o niveles de autoanticuerpos persistentemente elevados.
Pudieron evaluarse 6.605 pacientes (un 49% eran niñas, con una media de edad durante el seguimiento de 9 años [rango intercuartil: 8-10]). Desarrollaron anticuerpos positivos 1.216 (18%) y se confirmó la enfermedad celiaca en 447 (7%). El pico de mayor incidencia se encontró entre los 2 y los 3 años. El riesgo era mayor en los niños homocigotos para HLA DR3-DQ2. La ingestión de gluten se asoció de forma significativa con el riesgo de autoinmunidad positiva por cada aumento en 1 g en el consumo de gluten (Hazard ratio [HR]= 1,30; intervalo de confianza [IC] del 95%: 1,22-1,38); el riesgo absoluto a los 3 años si se consumía la cantidad de referencia de gluten era del 28,1%, pero si se consumía más de 1 g al día por encima del consumo de referencia, el riesgo absoluto ascendía al 34,2% (valor de la diferencia del 6,1%; IC del 95%: 4,5-7,7). Lo mismo ocurría respecto al riesgo de enfermedad celiaca: por cada gramo de aumento en el consumo de gluten aumenta el riesgo (HR= 1,50; IC del 95%: 1,35-1,65), un 20,7% de riesgo con el consumo basal, un 27,9% si el consumo era mayor de 1 g al día de gluten respecto al basal (diferencia de +7,2%; IC del 95%: 6,1-8,3).
Los autores concluyen que el consumo de altas cantidades de gluten en los primeros 5 años de vida en los niños con un riesgo genético aumentado se asocia a un riesgo mayor de desarrollar anticuerpos o enfermedad celiaca.
Nos quedan por conocer muchas cuestiones sobre la enfermedad celiaca y, por tanto, nos falta mucho para establecer recomendaciones consistentes de consumo de gluten para la población general: ¿cuándo debe incluirse en la dieta del niño?, ¿en qué cantidades? La incertidumbre es todavía mayor para las recomendaciones en familiares de primer grado de niños diagnosticados de enfermedad celiaca o diabetes mellitus. Quizá resultados como los de este estudio nos permitan aportar algún consejo para ellos, basado en datos científicos. Por ejemplo, restringir la cantidad de gluten en los primeros 5 años, en especial en los niños con mayor riesgo genético. Es cierto que los estudios longitudinales son laboriosos y costosos pero, sin duda, proporcionan una información relevante y permiten establecer hipótesis sólidas que deberán evaluarse con los estudios de intervención pertinentes.
Departamento de Pediatría. Clínica Universidad de Navarra. Madrid
Síndrome de apnea del sueño central en una lactante sin antecedente de prematuridad: ¿cuál es el manejo?
Granulomas hepatoesplénicos secundarios a infección por Bartonella. Presentación de un caso en una niña inmunocompetente y revisión de la literatura
Introducción: La bartonelosis es una causa frecuente de fiebre de origen desconocido con diversas formas de presentación.
Caso clínico: Exponemos el caso de una niña de 4 años que acude a urgencias por presentar fiebre de origen desconocido de 11 días de evolución. Durante su ingreso, presenta dolor abdominal periumbilical. En la ecografía abdominal se aprecian lesiones ocupantes de espacio hipoecogénicas hepatoesplénicas compatibles con granulomas, que se confirman en la tomografía computarizada abdominal. La familia refiere contacto casual con gatos en el domicilio. Se constata serología IgM positiva para Bartonella con seroconversión posterior. Recibe tratamiento con rifampicina y azitromicina durante 1 mes, con desaparición precoz de los síntomas y tardía de los granulomas.
Conclusión: El contacto epidemiológico con gatos, la sintomatología y los hallazgos ecográficos explicaron la afectación visceral de Bartonella. La mayoría de los casos descritos se dan en pacientes con alteración del sistema inmunitario. Sin embargo, esta entidad puede manifestarse en pacientes inmunocompetentes en los que el tratamiento es controvertido.
Consenso sobre la alimentación de los niños de 6-24 meses de edad: estudio ARMONÍA
Introducción: La alimentación del niño en sus 2 primeros años está influida por diversos factores que condicionarán su crecimiento y futura salud. Con este estudio se pretende elaborar un consenso sobre los factores implicados en la correcta alimentación de los niños de 6-24 meses de edad que pueda emplearse en la práctica clínica.
Material y métodos: Un comité científico de 3 pediatras expertos en nutrición infantil desarrolló una primera propuesta de ítems/dimensiones (fase I). La inclusión de estos ítems en el consenso fue evaluada por 51 pediatras. Se seleccionaron los ítems que alcanzaron acuerdo en ≥66,6% de los pediatras y/o ≥2 miembros del comité (fase II). Otros 29 pediatras más evaluaron el uso del consenso mientras atendían a 87 niños en su práctica clínica (fase III).
Resultados: Se alcanzó consenso en una sola ronda, que incluyó 33 de los 36 ítems propuestos, agrupados en 3 dimensiones: ambiente familiar (n= 6), historia clínica del niño (n= 4) y alimentación complementaria (n= 23). La evaluación del uso del consenso en la práctica clínica mostró que, en el 90,8-94,3% de los casos, los pediatras estaban conformes con los ítems y los comprendían satisfactoriamente. Además, el 70,1-88,5% lo consideró fácil de usar, con un tiempo de utilización adaptable al de la práctica clínica e integralmente adecuado.
Conclusiones: Este estudio permitió desarrollar un consenso sobre los factores a tener en cuenta en la correcta alimentación de los niños de 6-24 meses de edad que pueda emplearse como guía de consulta por los pediatras en su práctica clínica
Encuesta sobre conocimientos adquiridos en familias de recién nacidos con rasgo falciforme tras recibir educación sanitaria
Introducción: Los individuos portadores de rasgo falciforme (PRF) son personas sanas, asintomáticas. Esta afección implica la posibilidad de tener hijos con enfermedad de células falciformes y una serie de riesgos que deben conocerse.
Objetivo: Conocer el impacto educacional obtenido tras una primera consulta informativa sobre PRF.
Material y métodos: Estudio descriptivo transversal a través de encuestas telefónicas realizadas a los padres de hijos PRF que acudieron entre los meses de diciembre de 2014 a mayo de 2015 a la consulta tras un resultado positivo en las pruebas metabólicas para la anemia falcifome.
Resultados: De los 106 pacientes PRF sólo el 52,8% (56 personas) realizó la encuesta. Alrededor del 80% entendió la condición genética del rasgo falciforme y la posibilidad de tener hijos enfermos. El 55,4% entendió que la alteración en la hemoglobina era la responsable del rasgo falciforme, y 21 encuestados creían que el hemograma presentaba alteraciones secundarias al rasgo falciforme; el 48,2% había consultado con su pediatra tras el diagnóstico de PRF, mientras que el 44,6% de los encuestados no habían consultado o no se sentían satisfechos; el 76,8% entendió la importancia de informar de ser PRF en caso de una operación cardiaca; el 60,7% acudiría a un oftalmólogo en caso de traumatismo ocular con hemorragia; el 62% consideraba que había sido descartado cualquier otro tipo de anemia. Hasta 13 familias creían que ser PRF conllevaba un riesgo de desarrollar la enfermedad de células falciformes.
Conclusiones: Los resultados detectan llamativas deficiencias en la asimilación de la información por parte de las familias. Se impone la necesidad de mejorar el sistema de información, creando en primer lugar un grupo de trabajo que investigue las posibles causas.
Políticas públicas para reducir el consumo de bebidas azucaradas en niños y adolescentes / Pasar al menos 2 horas a la semana en contacto con la naturaleza se asocia con un mejor estado de salud y un mayor bienestar
Cada vez hay más datos de estudios epidemiológicos que sugieren que, a mayor contacto con la naturaleza (parques, bosques o playas), mejor es el estado de salud y el bienestar, al menos en poblaciones favorecidas, mayoritariamente urbanas. Vivir en una zona urbana más verde se asocia con una menor probabilidad de padecer enfermedad cardiovascular, obesidad, diabetes mellitus y enfermedad mental, entre otras afecciones, en población adulta, y con un menor riesgo de sufrir obesidad y miopía en niños. La mayoría de datos se obtienen de forma indirecta, a través del conocimiento de su lugar de residencia, pero no se correlaciona necesariamente con la cantidad de contacto «directo», es decir, el tiempo que realmente se pasa en ambientes naturales.
En este estudio los autores han diseñado un modelo para medir el tiempo real de contacto con un ambiente natural por semana y correlacionarlo con variables de salud bien conocidas. Como se trataba de una muestra nacional representativa, les permitió estratificar por variables socioeconómicas que podían condicionar los resultados. El estudio se realizó en Reino Unido y comprendía una muestra de 4.000 personas por semana, procedentes de las encuestas de los años 2014-2016 del programa «Monitor of Engagement with the Natural Environment» (MENE). La muestra final evaluada estaba constituida por 20.654 adultos. Tanto la valoración del estado de salud como del bienestar se realizaron mediante autodeclaración. La duración del contacto con el medio natural se obtuvo multiplicando el número de visitas semanales por la duración de una de las visitas escogidas aleatoriamente en esa semana. Con estos datos la duración se estratificó en 7 categorías (entre 0 y ≥300 min).
Los autores encontraron una asociación significativa entre los sujetos que señalaron que su estado de salud era bueno y el bienestar elevado con haber estado en contacto con la naturaleza ≥60 minutos/semana; esta significación se mantuvo después de corregir por las variables socioeconómicas sólo para el grupo que estuvo al aire libre ≥120 minutos/semana, sin encontrar una mejoría mayor con duraciones más prolongadas.
Por debajo de 120 minutos de visita semanal a ambientes naturales no se encontró ninguna diferencia al comparar con el grupo que no tenía ningún contacto, como si 120 minutos fuera un umbral a partir del cual se obtienen beneficios para la salud.
Entre las posibles explicaciones que los autores encuentran puede estar el hecho de que permanecer más tiempo en contacto con la naturaleza sea un marcador indirecto de actividad física. Señalan también que no importa el modo en que se llegue al umbral de 120 minutos para conseguir el beneficio (p. ej., largos paseos 1 o 2 veces a la semana frente a paseos diarios de corta duración).
Es cierto que el estudio tiene limitaciones, incluida la falta de evaluación de la «calidad» de la exposición; sin embargo, los hallazgos van en la línea de lo obtenido en los estudios epidemiológicos en que se estimaba de una forma indirecta las salidas al aire libre. Los autores sugieren en sus conclusiones que pasar ≥120 minutos a la semana en ambientes naturales puede representar un «umbral» importante para la salud y el bienestar en la población adulta inglesa.
A veces nos cuesta aceptar lo que parece de sentido común: vivir más en contacto con la naturaleza nos hace estar más sanos y sentirnos mejor. En una época en que parece que todo debe ser mensurable para ser creído y, por tanto, aceptado, sean bienvenidos estudios como el comentado para reforzar las ideas que proporcionan el sentido común y la observación. Como en muchas otras estrategias encaminadas a hacer que la población siga unos hábitos de vida saludables, la pregunta es cómo se lo transmitimos a los ciudadanos y, especialmente, cómo hacemos que nuestros niños y nuestros jóvenes aprecien que vivir más en contacto con la naturaleza no sólo consigue que se encuentren mejor hoy, sino que también tengan una vida más saludable y, ¿por qué no?, quizá más feliz.
Clínica Universidad de Navarra. Madrid
La alimentación en el niño: ¿es importante también cuándo come? Los ritmos circadianos en la alimentación infantil
En las últimas décadas se han publicado numerosos estudios observacionales en los que se muestra la relación que existe entre la desincronización de los patrones alimentarios y de sueño con el riesgo de padecer enfermedades metabólicas, en especial con la obesidad y la diabetes mellitus tipo 2. Los estudios realizados en niños son poco frecuentes. En esta revisión se comentan las bases de los mecanismos que regulan los ritmos circadianos en relación con la nutrición y las consecuencias de su alteración.
Dos estudios recientes muestran que los lactantes que hacen un mayor número de tomas durante la noche tienen un riesgo de tener un mayor índice de masa corporal durante la infancia. No se dispone de estudios en otros grupos de edad, excepto para la omisión del desayuno (asociada a un riesgo aumentado de padecer exceso de peso).
A pesar de la escasa evidencia científica disponible, parece razonable adecuar el patrón de alimentación (frecuencia de comidas, tipo de alimentos) a las necesidades variables a lo largo del día. Estas medidas pueden contribuir a mejorar el estilo de vida y a prevenir la enfermedad cardiovascular.
Advanced ultrasound techniques for pediatric imaging / Estado general de salud y satisfacción vital en niños con enfermedades crónicas
Advanced ultrasound techniques for pediatric imaging
Hwang M, Piskunowicz M, Darge K. Pediatrics. 2019; 143: e20182609.
Resumen
La ecografía constituye una herramienta esencial en el abordaje diagnóstico en pediatría, por su disponibilidad, seguridad y bajo precio. En esta revisión se presentan los avances más recientes en la técnica ecográfica y su aplicabilidad clínica, que se concretan en mejoría de la sensibilidad de la escala de grises convencional, el uso de Doppler color y la posibilidad de obtener una información funcional adicional. Además, la revisión ilustra el empleo de cada técnica en casos clínicos paradigmáticos.
Ecografía de contraste
En este tipo de ecografía se usan agentes de contraste que son microburbujas de gas microencapsuladas con fosfolípidos de 2-3 µm de tamaño, que generan una señal potente de ultrasonidos. La técnica requiere la inyección del contraste en una vía periférica, que se elimina por exhalación a través de los pulmones, por lo que es más seguro que el contraste usado en la tomografía computarizada (TC) o en la resonancia magnética (RM). Además, elimina la necesidad de sedación en niños.
La principal utilidad de esta prueba en niños pequeños es su capacidad para diagnosticar algunas lesiones focales benignas en el hígado, como los hemangiomas, la hiperplasia nodular o la infiltración grasa. Al ser capaz de delimitar bien el patrón de vascularización de una lesión, puede ser de ayuda para distinguir entre lesiones benignas y malignas. En las lesiones malignas, generalmente muy vascularizadas, el fenómeno de aclarado rápido del contraste es uno de los rasgos más característicos de la ecografía de contraste y, además, puede identificar áreas de necrosis frente a áreas de tumor viable, por lo que sirve de guía para las biopsias dirigidas. Como es útil para el seguimiento de estas lesiones, disminuye la necesidad de realizar TC o RM.
También es útil para el diagnóstico de la isquemia o el daño en un órgano (p. ej., para valorar el grado de afectación en una enterocolitis necrosante). En el caso de los lactantes podría incluso servir como método alternativo para el diagnóstico de muerte cerebral. También serviría para valorar el grado de inflamación, fibrosis o cicatrización (p. ej., para distinguir lesiones fibróticas o una inflamación en un paciente con enfermedad de Crohn).
Una indicación de esta técnica aprobada por la Food and Drug Administration en niños es su empleo en la urosonografía de eliminación, para el diagnóstico del reflujo vesicoureteral, como una alternativa a la cistourografía convencional. La técnica consiste en la administración de las microburbujas a través de una sonda urinaria y evaluar la existencia de reflujo con la ecografía. Su mayor sensibilidad y la posibilidad de evaluar de forma detallada la uretra han posibilitado que reemplace a la cistografía convencional.
Elastografía
La elastografía es una técnica de imagen funcional que valora la rigidez de los tejidos determinando la velocidad de conducción de las ondas de ultrasonido. Cuanto más rígido es un tejido, más rápido viajan las ondas.
Hay dos tipos de elastografías: de onda cortante (hear-wave) y de tensión o de deformación (strain). La primera se usa para cuantificar el grado absoluto de rigidez de un tejido, y se basa en aplicar ondas de alta intensidad midiendo cómo se propagan lateralmente con la deformación de los tejidos. La segunda es una técnica semicuantitativa de la compresión manual o el movimiento interno fisiológico (p. ej., el latido cardiaco) que resultan en el desplazamiento axial reflejo de la elasticidad del tejido.
Una de las principales ventajas de la elastografía es que ha disminuido la necesidad de realizar biopsias hepáticas. Podría usarse en el diagnóstico de otras patologías, como la detección de un segmento fibrótico en el intestino o de cicatrices renales.
Ecografía en 3 o en 4 dimensiones
Tradicionalmente, el inconveniente de las ecografías en 3 dimensiones (3D) era la duración del procesamiento de las imágenes. Los nuevos avances tecnológicos permiten realizar reconstrucciones de imágenes muy rápidas a partir de los cortes en 2 dimensiones. Son útiles para determinar el volumen de un órgano o de una masa. La ecografía en 4D permite añadir la dimensión tiempo a la ecografía en 3D.
Ecografía Doppler ultrarrápida
Consiste en una técnica Doppler avanzada con una resolución muy alta, muy superior a la convencional. Permite detectar con una sensibilidad 50 veces superior los cambios de flujo en el cerebro, que se correlacionan bien con la actividad neuronal valorada en el electroencefalograma. Esta resolución temporal es muy superior a la de la RM funcional.
Ecografía de alta frecuencia
La ecografía de alta frecuencia proporciona un gran aumento en la resolución, hasta llegar a los 30 µm, superior a la TC o la RM. Se aplica principalmente en neonatología y dermatología, o para el estudio neuromuscular, vascular de tiroides, linfáticos, etc.
Lo que aporta este estudio:
La ecografía ya constituía una herramienta clave en el diagnóstico de todo tipo de lesiones en el paciente pediátrico. Los avances tecnológicos permiten aumentar mucho sus posibilidades no sólo para el diagnóstico, sino también para el seguimiento de las lesiones y, por tanto, como guía en el tratamiento. Ya no sólo proporcionan imágenes, sino que además aportan una información funcional. La ecografía combina facilidad de desplazamiento, bajo precio, ausencia de radiación y sedación innecesaria junto con seguridad, por lo que adquiere una gran relevancia como herramienta en la clínica. Quizás, en un sistema como el español, haya que dilucidar la accesibilidad de esta técnica y la dependencia del radiólogo para su realización. Mientras que otras técnicas de imagen pueden realizarlas los técnicos ante de ser evaluadas por los radiólogos, de momento no ocurre lo mismo con la ecografía. Probablemente, en el marco de la profesión se requiera una reflexión sobre el mejor modo de hacer accesible a la población infantil una herramienta de diagnóstico con tantas virtudes y tan pocos inconvenientes como la ecografía.
J.M. Moreno-Villares
Clínica Universidad de Navarra. Madrid
Estado general de salud y satisfacción vital en niños con enfermedades crónicas
General health and life satisfaction in children with chronic illness
Blackwell CK, Elliott AJ, Ganiban J, Herbstman J, Hunt K, Forrest CB, et al. General health and life satisfaction in children with chronic illness. Pediatrics. 2019; 143(6): e20182988.
Resumen
Los autores de este artículo se plantearon investigar cómo repercuten las enfermedades crónicas en el estado general de salud y en la satisfacción vital de los niños. Gracias a los avances de la medicina y a los cuidados específicos, algunas enfermedades que hace un tiempo eran mortales son actualmente tratables. Se incrementa cada vez más la expectativa de vida, por lo que también se eleva el número de años que se vive con esas enfermedades. La prevalencia de enfermedades crónicas en Estados Unidos y en otros países desarrollados continúa en aumento. Se estima que 1 de cada 5 niños tiene una enfermedad crónica. Los pacientes con estas enfermedades pueden sufrir tanto limitaciones físicas y retrasos del desarrollo psicomotor como dificultades escolares y sociales. De todas formas, aún no se ha determinado el grado de satisfacción vital que tienen estos niños a pesar de su enfermedad.
La satisfacción vital es un componente de la salud y parte de la construcción multidimensional del bienestar, y se define como la evaluación individual de la propia vida como buena y satisfactoria.
Es llamativa la ausencia de estudios sobre satisfacción vital en el contexto de enfermedades crónicas, especialmente en la edad pediátrica.
Los investigadores de este trabajo plantearon dos hipótesis: a) la enfermedad crónica está negativamente asociada con la salud general, y b) la enfermedad crónica no se asocia con la satisfacción vital.
En total, 1.113 cuidadores, pertenecientes a 3 estudios simultáneos de cohortes, completaron el Patient-Reported Outcomes Measurement Information System (PROMIS) para padres con el fin de evaluar el estado general de salud física, mental y social, así como el PROMIS en relación con la satisfacción vital. Participaron 1.253 niños de 5-9 años de edad, entre marzo de 2017 y diciembre de 2017. Un 20% de los niños tenía, al menos, una enfermedad crónica, y un 8,5% más de dos. Los investigadores armonizaron los factores demográficos y los factores ambientales y familiares estresantes (familias monoparentales, salud mental materna e ingresos familiares) con medidas comunes en todas las cohortes. Para examinar las asociaciones entre enfermedades crónicas y el estado de salud de los niños y la satisfacción vital, se ajustaron modelos de regresión lineal.
En cuanto a los resultados del estudio, como era de esperar, los niños con enfermedades crónicas tenían un peor estado general que aquellos sin enfermedades (β ajustada= –1,20; intervalo de confianza [IC] del 95%: –2,49 a 0,09). Sorprendentemente y por contraste, los niños con enfermedades crónicas tenían similares niveles de satisfacción vital (β ajustada= –0,19; IC del 95%: –1,25 a 0,87). En cambio, los niños con diagnóstico psicológico de estrés tenían la asociación negativa más fuerte, tanto para el estado de salud como para la satisfacción vital.
Comentario
La plataforma PROMIS se creó en 2004 por el National Institutes of Health de Estados Unidos para poder evaluar el estado de salud y el resultado de las intervenciones que se realizan al respecto. Es un sistema de información que mide los resultados referidos por los propios pacientes. La valoración del estado de salud, especialmente el de la satisfacción vital, es algo muy personal. Para los investigadores es un reto saber qué es lo que sucede en el interior de los pacientes, cómo valoran su estado de salud y su calidad de vida, y hasta qué grado son consistentes sus respuestas. Esta preocupación ha llevado a la creación de múltiples herramientas de medida del estado de salud y de las intervenciones sociosanitarias. Así surge PROMIS como resultado del trabajo colaborativo de grupos de investigación y centros de reconocido prestigio estadounidenses. Esta plataforma permite generar medidas de resultados referidos por los pacientes de forma estandarizada. Además, se ha hecho el esfuerzo de adaptarlas idiomáticamente para que su uso pueda tener un alcance internacional en la investigación y la práctica clínica. Durante el periodo 2006-2007, el PROMIS recopiló datos de muestras amplísimas, tanto de pacientes como de población general americana, asegurando que existiera una representación adecuada según la edad, el sexo, el nivel educativo, el estatus socioeconómico y el grupo étnico. Con toda esta información se crearon «bancos de ítems», que son herramientas muy útiles para medir aspectos relacionados con la calidad de vida. La plataforma PROMIS cuenta además con el apoyo de modelos matemáticos, que permiten una mayor precisión a la hora de valorar las respuestas de los pacientes. Con estos análisis matemáticos se puede medir el funcionamiento mental humano a la hora de responder a un cuestionario sobre su salud, estableciendo una relación entre el nivel de habilidad del examinado y la probabilidad de que responda correctamente a las preguntas. Los resultados obtenidos son más fiables y se simplifica el estudio, ya que de esta manera los investigadores no necesitan un tamaño muestral tan grande, al contar con herramientas ya validadas y de aplicación universal.
Los autores de este artículo concluyen que, aunque los padres de los niños con enfermedades crónicas valoran con peores resultados la salud de sus hijos, la satisfacción vital de estos niños resulta comparable con la de sus compañeros sanos.
Los niños con enfermedades crónicas aprenden a afrontar y superar situaciones adversas (dolor, sufrimiento, limitaciones funcionales...) y nos dan una lección a los adultos cuando les vemos sonreír y jugar a pesar de sus dificultades.
Lo que aporta este artículo:
Este trabajo de investigación tiene el interés de haber sido realizado a través de la plataforma PROMIS, considerada una herramienta útil y fiable para estudios en relación con el estado de salud y la satisfacción vital, también en la edad pediátrica.
Los progresos de la medicina están facilitando que sobrevivan cada vez más niños con enfermedades graves y crónicas. Con este estudio los autores aportan la evidencia de que las enfermedades crónicas no imposibilitan a los niños tener una vida feliz y satisfactoria.
C. Esteve Cornejo
Pediatra. Clínica Universidad de Navarra. Madrid
Cibermetría y pediatría: una relación manifiestamente saludable (XV)
El principal objetivo de este trabajo es mostrar cómo la correcta aplicación de técnicas cibermétricas a la pediatría puede proporcionar una ingente cantidad de información que, correctamente analizada y contextualizada, facilita la toma de decisiones estratégicas a los distintos agentes involucrados de forma directa o indirecta en esta especialidad médica. En el presente artículo se proponen dos análisis complementarios. El objetivo del primer análisis es determinar la presencia online de las sociedades españolas de pediatría. Para ello, se recopilaron diversas métricas online relacionadas con la presencia, la visibilidad, el impacto y la conectividad de 39 sociedades. Los resultados indican que las sociedades españolas de pediatría no disponen de sitios web de gran impacto y que utilizan principalmente Facebook y Twitter, aunque el uso de estas plataformas difiere ampliamente entre ellas, lo que refleja la existencia de distintas estrategias de comunicación web. El objetivo del segundo análisis es evaluar el impacto de los hashtags relacionados con la vacunación en Twitter, para lo cual se analizaron hashtags en español relacionados con las vacunas sistemáticas recomendadas por la Asociación Española de Pediatría (un total de 18). Los resultados muestran un bajo volumen de contenidos (pocos tweets), que despiertan escaso interés (pocos comentarios) y que obtienen una difusión (retweets) e impacto (likes) moderados.
Automedicación en una muestra de adolescentes y su asociación con el consumo de tabaco, alcohol y la calidad de vida relacionada con la salud
Introducción: El inicio del consumo de tabaco y alcohol y el descenso de la calidad de vida relacionada con la salud (CVRS) se producen mayoritariamente en la adolescencia, etapa en la que se desarrolla la automedicación. El objetivo del presente estudio es describir estas variables y explorar las diferencias por sexo y edad.
Sujetos y métodos: Estudio descriptivo transversal de una muestra de adolescentes. Se aplicó una escala de CVRS y cuatro indicadores de salud sobre el uso de medicamentos, tabaco y alcohol. Se utilizó el análisis de la varianza (ANOVA) para comprobar las diferencias entre sexos y edades en los consumos, el análisis multivariante de la varianza (MANOVA) para la CVRS y las correlaciones de Pearson para explorar las relaciones entre las variables.
Resultados: La tasa de consumo de analgésicos/antipiréticos fue del 65,1%, antibióticos del 40% y tranquilizantes/relajantes/para dormir del 9%, de los que se consumieron sin prescripción el 59,1, el 33 y el 63,3%, respectivamente. En el ANOVA se hallaron diferencias estadísticamente significativas entre sexos para el uso de medicación, y entre edades para el resto de los indicadores. El MANOVA mostró también diferencias en la CVRS según el sexo y la edad. Todas las correlaciones estadísticamente significativas (a excepción de la existente entre el «apoyo social y pares» y el «consumo de alcohol») fueron negativas.
Conclusiones: Las chicas presentan una peor CVRS y una mayor automedicación, y los adolescentes de 17 años una peor CVRS y un mayor consumo de medicamentos, tabaco y alcohol. Las correlaciones entre la CVRS y los indicadores son predominantemente negativas. Se encuentran correlaciones entre las dimensiones de CVRS y el consumo de sustancias, diferenciadas según el sexo y la edad.
Evaluación de la actividad física en niños
La práctica de actividad física desempeña un papel fundamental en el desarrollo y la salud del niño, así como en la lucha contra la obesidad infantil. Esta revisión tiene como objetivo describir los diferentes métodos de evaluación de la actividad física para poder utilizarlos en investigación y, especialmente, en la práctica clínica. Existen métodos objetivos y subjetivos de valoración, entre los que destaca la acelerometría como método de referencia. El acelerómetro es un dispositivo que se coloca a la altura de la cadera, sujeto por un cinturón, que registra la duración y la intensidad de los movimientos que realiza un niño a lo largo de un día. Por otro lado, el uso de podómetros o pulseras de actividad es una alternativa más económica para realizar medidas de forma objetiva, aunque tienen menor precisión. Entre los métodos subjetivos, los cuestionarios y diarios de actividad física destacan por su fácil accesibilidad y reproducibilidad, así como por una amplia comparación con otros estudios. Entre los descritos en la literatura, hay algunos más indicados para la población infantil, como el cuestionario Assessment of Physical Activity Levels Questionnaire (APALQ). Por último, algunas técnicas, como el agua doblemente marcada, la calorimetría o la observación directa, son métodos de difícil acceso para el pediatra y de elevado coste económico, aunque ya se están utilizando en algunos centros especializados y en investigación. En conclusión, todas estas herramientas pueden permitir al pediatra prescribir actividad física y evaluar su práctica con mayor precisión para poder individualizar el tratamiento en función de las características del niño y su cumplimiento.
Falsos mitos sobre alergias alimentarias

Nutribén® esclarece las 5 creencias más comunes entre los padres
- Enfermedad celíaca. Es una Intolerancia permanente al gluten. Esta requiere de una alimentación estricta sin presencia del gluten, ya que es la única terapia existente para sobrellevarlo.
- Sensibilidad al gluten, no celíaca. A diferencia de la enfermedad celíaca, esta puede ser transitoria y desaparecer al cabo de un par de años sin la ingesta de gluten.
- Alergia al trigo. Esta sí que se presenta como un caso de alergia al cereal al 100%, con todos los riesgos que ello supone.
1. Clasificación de la Academia Europea de Alergia e Inmunología clínica.
2. Estudio de la Sociedad Española de Inmunología Clínica, Alergología y Asma Pediátrica.
Los cereales en la alimentación del lactante y el niño pequeño
Protocolo de actuación en atención primaria en la enfermedad inflamatoria intestinal en la edad pediátrica
Escoliosis idiopática en adolescentes / Disciplina eficaz para criar niños sanos
Caracterización del desarrollo en niños con síndrome de Down de 5-12 años de edad en Bogotá (Colombia)
Efectos del juego en el desarrollo infantil (I): aspectos teóricos
Este artículo tiene por objetivo principal describir cómo el juego puede ejercer una influencia importante en la mejora del aprendizaje infantil. En este sentido, el juego no sólo se considera un acto espontáneo entre niños y niñas, sino también una actividad dirigida, que puede utilizarse como recurso en todas las etapas educativas e, indiscutiblemente, en los diferentes entornos en que se desenvuelve la población infantil: la familia, el barrio, los espacios de ocio, deportivos, etc. En la elaboración de este artículo se ha seguido un método de revisión bibliográfica, cuyos resultados muestran los efectos que puede tener el juego sobre los niños, en todas sus variantes; asimismo, se presentan algunas sugerencias para su gestión y control. Se pretende dar continuidad a esta revisión bibliográfica con un próximo artículo, en el que se intentará realizar un estudio de campo sobre el impacto de los juegos digitales en niños de diferentes franjas de edad.
Valoración subjetiva de los padres en el manejo de las bronquiolitis
Programa de transición de nefrología pediátrica a la medicina del adulto: «Conduce tu salud»
El otoño abarca el 20% de las visitas de urgencia en Pediatría

- Entre las causas principales se encuentran aquellas relacionadas con infecciones de las vías respiratorias, las alergias y el aparato digestivo
- La microbiota tiene un papel clave en el desarrollo de un sistema inmunológico fuerte y la prevención de enfermedades
- Los prebióticos y probióticos contribuyen a mantener el equilibrio en la microbiota intestinal y reforzar el sistema inmunitario de pequeños y adultos
Luxación congénita de rótula. Diagnóstico en un adolescente sano
La luxación congénita de rótula es una entidad clínica excepcional, en la que la rótula se encuentra luxada de forma permanente y no puede reducirse de forma manual. Se suele manifestar al nacimiento, aunque en algunos casos el diagnóstico puede retrasarse hasta la adolescencia o la edad adulta. El diagnóstico temprano permite la corrección quirúrgica con el fin de evitar las secuelas tardías, incluidos los cambios degenerativos prematuros de la rodilla. Se presenta un caso de luxación congénita de la rótula en un adolescente de 12 años de edad, sano y jugador de fútbol.
Bibliometría e indicadores de actividad científica (X). Indicadores cienciométricos en Scimago Journal and Country Rank. Análisis de la categoría temática «Pediatrics, Perinatology and Child Health»
En este artículo se describe el portal Scimago Journal and Country Rank, creado por el grupo de investigación español Scimago, que calcula una serie de indicadores métricos de impacto obtenidos a partir del análisis de las revistas indexadas en la base de datos Scopus. Se describen las cuatro posibles opciones de consulta, tanto el análisis específico de una revista de forma individual como a través de la comparación de revistas (Journal Ranks), la clasificación de los países evaluada según su producción científica (Country Ranks), así como una nueva herramienta de visualización de la información (Viz Tools). Los resultados obtenidos utilizando como ejemplo el análisis de la revista Acta Pediátrica Española proporcionan, junto con la información bibliográfica de la revista, las métricas Scimago Journal Rank (SJR), el índice H, los documentos, las referencias, las citas, las citas por documento y el país, todo ello evaluado anualmente para el periodo 1999-2016.
Sobrepeso y obesidad en adolescentes adoptados internacionalmente
Derivaciones para la realización de ecografías en pediatría de atención primaria: utilidad de una guía de indicaciones clínicas
Meningitis y responsabilidad profesional médica en pediatría: ¿en qué podemos mejorar?
La Cátedra Ordesa reúne en Barcelona a destacados expertos internacionales en nutrición infantil

Jornadas Científicas de la Cátedra Ordesa de Nutrición Infantil
Los primeros 1.000 días de la vida de una persona pueden condicionar su salud durante el resto de su vida. Algunas evidencias científicas muestran que la nutrición y el estilo de vida en los primeros años pueden programar nuestra salud y el riesgo de padecer enfermedades en el futuro. De esta forma, la nutrición infantil se ha convertido en una de las principales áreas de investigación actuales.
La Cátedra Ordesa de Nutrición Infantil abordará en sus V Jornadas Científicas Internacionales, que se celebrarán los próximos días 26 y 27 de abril en Barcelona, los últimos avances científicos que guiarán el futuro de la alimentación infantil desde los primeros meses de vida, para conseguir un envejecimiento más saludable y contribuir a prevenir enfermedades.
Las Jornadas Científicas Internacionales están organizadas por la Cátedra Ordesa de Nutrición Infantil, impulsada por Laboratorios Ordesa y ubicada en la Universidad de Granada y cuenta con la colaboración de la Universidad de Zaragoza y la Universidad de Cantabria. El encuentro reunirá más de 300 especialistas en Pediatría procedentes de más de 10 países de Europa, América, Oriente Medio y Asia.
Entre los ponentes asistentes destacan: Cristina Campoy, directora de la Cátedra Ordesa de Nutrición Infantil y Titular del Departamento de Pediatría de la Facultad de Medicina de la Universidad de Granada, miembro de la Sociedad Europea de Gastroenterología, Hepatología y Nutrición Pediátricas (ESPGHAN) y la Early Nutrition Academy (ENA); Alessandro Fiocci, director del Departamento de Alergías en el Hospital Pediátrico Bambino Gesú de Roma; Francisco Guarner, Científico principal de la Unidad de Investigación de Sistema Digestivo del Institut de Recerca Vall d’Hebrón de Barcelona y Presidente de la Sociedad Española de Probióticos y Prebióticos (SEPYP); Olivier Goulet, profesor de Pediatría en la Universidad de París V-René Descartes y Presidente del Centro Nacional de Referencia para Enfermedades Raras del Necker University; Marjo-Riita Jarvelin, Catedrática en Epidemiología de Trayectoria Vital en el Imperial College de Londres; Luis Moreno, Catedrático de la Facultad de Ciencias de la Salud de la Universidad de Zaragoza, Coordinador del Grupo de Investigación GENUD (Growth, Exercise, Nutrition and Development) de la Universidad de Zaragoza y Presidente de la Sociedad Española de Nutrición (SEN); Susan Ozanne, Profesora de Desarrollo Endocrino de la Universidad de Cambridge y miembro del Consejo de la International Society for the Developmental Origin of Health and Disease; Hanja Szwajeska, Doctora y Catedrática del Departamento de Pediatría de la Universidad de Varsovia (Polonia) y Editora Jefe del Journal of Pediatric Gastroenterology and Nutrition; e Yvan Vandenplas, Catedrático en Pediatría y Jefe del Hospital Pediátrico KidZ Health Castle en el Hospital Universitario de Bruselas.
V JORNADAS CIENTÍFICAS DE LA CÁTEDRA ORDESA DE NUTRICIÓN INFANTIL
Día:
La importancia de la nutrición en los primeros 1.000 días de la vida
Contaminación ambiental e ingresos pediátricos en un área urbana
La recomendación de lactancia materna exclusiva hasta los 6 meses de vida: algunas consideraciones
La recomendación global de la Organización Mundial de la Salud (OMS) de dar «lactancia exclusivamente materna durante los primeros 6 meses de vida» es considerada por las madres y los profesionales sanitarios un objetivo nutricional idóneo para cualquier lactante, si así se desea y no hay impedimento para efectuarla. Es importante destacar que el llamado «Tercer Mundo», con millones de desfavorecidos, era y es el objetivo fundamental de la difusión y puesta en marcha de un programa de salud de la OMS muy amplio, en el que se incluye dicha recomendación. Las circunstancias socioeconómicas y culturales en los países de nuestro ámbito occidental son diferentes, con una historia bien establecida, por lo que las recomendaciones de la European Society for Paediatric Gastroenterology, Hepatology and Nutrition son compatibles con la citada recomendación de la OMS respecto a la introducción de la alimentación complementaria desde los 4 meses. Por ello, esta recomendación debe aplicarse con flexibilidad en nuestro ámbito pediátrico y vigilar los riesgos de ferropenia que puede conllevar.
Pfizer presenta Trumenba®, la nueva vacuna frente al meningococo B diseñada para ofrecer amplia cobertura frente a la mayoría de cepas circulantes

- Más del 90% de las cepas circulantes de meningococo B en España estarían cubiertas con la nueva vacuna Trumenba®
- Trumenba® puede administrarse a partir de los 10 años de edad y junto con otras vacunas del calendario oficial
- Trumenba® tiene un perfil de seguridad basado en once estudios clínicos en todo el mundo, con más de 15.000 sujetos de 10 años de edad y mayores
Alteraciones auditivas y desarrollo del lenguaje en niños con síndrome de Down: revisión sistemática de la literatura
¿Seguimos considerando el tabaquismo pasivo como un problema de salud?
Banco de sangre del cordón umbilical para un posible trasplante futuro / La exposición a un ambiente verde en el lugar de residencia
Cord blood banking for potential future transplantation
Pediatrics. 2017; 140(3): e20171870.
Centro de Salud «María Montessori». Leganés (Madrid)
Éste es el objetivo que pretenden los autores del trabajo comentado, basado en los resultados del seguimiento de dos cohortes de niños de Sabadell y Valencia valorados desde su nacimiento hasta los 7 años. Un medio ambiente natural proporciona a los niños oportunidades únicas de compromiso, descubrimiento, medida del riesgo, creatividad, control y fortalecimiento del autodominio, además de inspirar emociones básicas (como la capacidad de asombro), factores que influyen de una forma positiva en el desarrollo cognitivo y en la atención.
Además, de una forma indirecta, influyen favoreciendo la actividad física, disminuyendo la exposición a la polución ambiental y a los ruidos y contribuyendo al contacto con un ambiente rico en microbios.
Algunos estudios previos habían demostrado que la exposición de niños de educación infantil a un «ambiente verde» (surrounding greenness) se asociaba a una mayor capacidad de memoria y atención. Sin embargo, a la edad de la exposición (7-10 años) ya existe un desarrollo cognitivo sustancial, por lo que no fue posible evaluar las influencias en las etapas de rápido desarrollo cerebral, como la prenatal o la posnatal inmediata.
Con el fin de valorar esas influencias tempranas, era de mayor interés realizar el seguimiento de una o varias cohortes desde el nacimiento y medir la atención durante la etapa preescolar y a los 7 años de edad.
En el trabajo comentado, la exposición a la naturaleza se evaluó mediante dos índices de verdor basados en imágenes de satélite (teledetección): índice normalizado de vegetación diferente (normalized difference vegetation index [NDVI]) e indicador de cubierta vegetal (vegetation continuous fields [VCF]). Ambas medidas se realizaron en las distancias de 100, 300 y 500 metros de la vivienda habitual, en distintos periodos hasta los 7 años de edad. La valoración de la atención se efectuó mediante pruebas específicas automatizadas (Conners’ Kiddie Continuous Performance Test y Attentional Network Task), que permiten evaluar los errores de omisión o de acción y la velocidad de respuesta.
La evaluación se pudo realizar en 1.199 niños a los 4-5 años de edad y en 1.044 a los 7 años. Los resultados mostraron que los niños con más exposición a un ambiente verde cometían menos errores de omisión. Estos resultados se mantenían cuando se tenían en cuenta ciertas variables, como el número de hermanos y el orden en la fratría, haber sido o no amamantados, el peso al nacimiento o el número de horas delante del televisor o de cualquier pantalla, la clase social y el nivel educativo de las madres.
La exposición a un ambiente verde en el lugar de residencia se asoció de forma significativa a una menor tasa de errores de omisión y a un tiempo de respuesta más corto, que podría interpretarse como una mayor capacidad de atención dirigida en ese grupo de niños. Los resultados fueron más positivos cuando se evaluaba la presencia de áreas verdes cercanas al domicilio que cuando se evaluaba la cubierta vegetal (presencia de árboles).
Lo que aporta este estudio:
Los seres humanos tienen unos lazos evolutivos importantes con la naturaleza. Así, el contacto con la naturaleza se asocia a un mejor desarrollo cerebral en los niños; en especial, favorece la atención. La capacidad de asombro es una característica del ser humano que promueve el desarrollo de aptitudes y habilidades. Y la naturaleza es una fuente inagotable, aunque no la única, de asombro. Fomentar la creación de espacios verdes en los entornos urbanos, el acercamiento de los niños a la naturaleza y las actividades al aire libre (sobre todo el juego) son elementos clave para conseguir no sólo que los niños sean más inteligentes, sino probablemente más felices.
Los cultivos de leche humana 5 años después de la primera propuesta de protocolo (I): una herramienta útil para la salud pública
Análisis de la metodología «trigger tools» en el servicio de urgencias de un hospital terciario
Prevención integral de la obesidad y sobrepeso en niños preescolares
Resultados de la campaña sanitaria «Peso y salud infantil» en las farmacias comunitarias de la provincia de Cádiz
Fiebre-fobia nosocomial: un miedo contagioso
Efectos de la acupuntura mínima para los cólicos infantiles / Estudio INSIGHT: repercusiones en el sueño cuando madre y niño comparten habitación
Mother-infant room-sharing and sleep outcomes in the INSIGHT study
Frecuentación en urgencias de los neonatos en un área suburbana de Madrid
El 78% de los casos de bebés con bronquiolitis aguda (BA) grave son causados por el VRS
Primer estudio prospectivo multicéntrico en España realizado en 16 Unidades de Cuidados Intensivos Pediátricos (UCIP) españolas sobre epidemiología y manejo actual de la bronquiolitis aguda grave
- El estudio ha sido coordinado por el Dr. José C. Flores-González, de la UCIP del Servicio de Pediatría del Hospital Universitario Puerta del Mar de Cádiz, con el apoyo del Hospital Sant Joan de Déu de Barcelona y Complejo Hospitalario de Pamplona y la participación de un total de 16 hospitales de toda España con 208 lactantes incluidos.
- El 26,7% de los bebés ingresados en esas UCIP presentaban factores de riesgo de una bronquiolitis aguda grave, siendo ésta la principal causa de ingreso hospitalario en bebés menores de un año1,2.
Leches de crecimiento. ¿Qué pueden aportar en la alimentación del niño pequeño?
Inmigración y riesgo de sobrepeso y obesidad en niños en edad escolar
La pediatría en las Jornadas Internacionales de Cine Médico de San Sebastián
Novedad en alimentación infantil: fórmulas a base de leche de cabra
Morbimortalidad en recién nacidos con una edad gestacional igual o inferior a 26 semanas: estudio de los límites de viabilidad en nuestro medio
Introducción: La toma de decisiones acerca de los cuidados perinatales en fetos y recién nacidos (RN) al límite de la viabilidad continúa siendo un problema clínico y ético de la máxima envergadura para obstetras y neonatólogos. La inclusión de los padres en el proceso de toma de decisiones exige que la información proporcionada esté basada en los mejores datos disponibles. El objetivo de nuestro estudio fue conocer las tasas específicas de supervivencia al alta y de supervivencia sin morbilidad mayor, por edad gestacional (EG), en RN ≤26 semanas.
Pacientes y métodos: Durante el periodo 2004-2010 se recogieron datos de todos los RN vivos (RNV) intramuros, de ≤26 semanas de EG. Se estudiaron los datos demográficos, de intervenciones y los resultados en morbimortalidad específica por EG.
Resultados: Se incluyeron 137 RNV con una EG ≤26 semanas, de los que 8 fallecieron en la sala de partos. Entre los 129 ingresados en la unidad de cuidados intensivos neonatales, la supervivencia se incrementó progresivamente en función de la EG desde el 30,4% a las 24 semanas hasta el 64,7% a las 26 semanas. Asimismo, la supervivencia sin morbilidad mayor se incrementó desde el 4,3 al 25,9% en dichas EG. La mediana (rango intercuartílico) de la estancia hospitalaria en los supervivientes fue de 90 (76,5-113) días, y en los que fallecieron de 8 (3-21,5) días.
Conclusiones: La supervivencia y la supervivencia sin morbilidad mayor aumentan significativamente con la EG en RN al límite de la viabilidad. Estos resultados, específicos por EG, aportan una información relevante para la toma de decisiones asistenciales e indican el potencial impacto en la gestión de recursos sanitarios.
Top 5.000 en el blog «Pediatría basada en pruebas» y el interés de la «blogmetría»
Los efectos del consumo de leche y derivados lácteos sobre la salud / Gestión de las dietas en las alergias alimentarias
Curr Opin Pediatr. 2016; 28: 567-572.
Junto a la bien tipificada alergia mediada por IgE, en la última década se ha avanzado en el conocimiento y la detección de alergia a alimentos no mediada por IgE (son buenos ejemplos de ella el síndrome de enterocolitis inducida por proteínas alimentarias, la enteropatía inducida por alimentos, la protocolitis o la esofagitis eosinofílica). Ya sea uno u otro tipo, conllevan el requerimiento de una dieta restrictiva.
Cuando la restricción abarca varios grupos de alimentos, puede verse comprometido el crecimiento o la ingesta de determinados nutrientes, como el calcio y la vitamina D. Cuanto mayor es el número de alimentos prohibidos, mayor es el riesgo de que se presenten deficiencias nutricionales, de los que en este artículo se comentan algunos casos paradigmáticos (deficiencia de yodo, déficit de ácidos grasos esenciales, xeroftalmia por deficiencia de vitamina A o raquitismo por déficit de vitamina D).
En el seguimiento de los pacientes con alergias alimentarias es preciso un abordaje multidisciplinario. Deben recibir información y consejo sobre los alimentos que hay que evitar, dónde pueden estar presentes y la interpretación del etiquetado, así como sobre las alternativas para alcanzar los requerimientos o cuándo y qué tipo de suplementos se pueden utilizar. Las familias de estos niños deben conocer también los planes de provocación/introducción de los alimentos prohibidos. Se revisa también la recomendación sobre la vacuna de la gripe en niños con alergia al huevo, y se recomienda el uso de la vacuna intranasal o las vacunas inactivadas no basadas en cultivos. No hay ninguna contraindicación para usar la vacuna triple vírica.
Los autores revisan también el estado actual de la inmunoterapia oral de desensibilización y otras alternativas, como la inmunización epicutánea. Finalmente, hacen una reflexión sobre los resultados de estudios recientes (LEAP, con introducción muy precoz en la dieta de alimentos potencialmente alergénicos), que obligarán en un futuro próximo a la actualización de las guías de introducción de la alimentación complementaria.
Los pediatras se enfrentan cada día a más casos y nuevas formas de presentación de la alergia alimentaria. Surgen nuevos métodos diagnósticos, alternativas de tratamiento y medidas de prevención. En la práctica, una alergia alimentaria cambia la vida diaria del niño y su familia. El profesional encargado de su atención debe anticiparse a los posibles problemas y buscar el apoyo de servicios de referencia para los casos más complejos. ¡Ah!, y habrá que estar atentos a las nuevas recomendaciones sobre el momento y la forma de introducir los alimentos en la dieta del lactante.
Factores de riesgo de la mastitis infecciosa durante la lactancia
La mastitis infecciosa es una patología común durante la lactancia y constituye una de las primeras causas de destete precoz. Por tanto, debería considerarse un problema de salud pública relevante, ya que priva a la pareja madre-hijo de los incuestionables beneficios de la lactancia. No obstante, la mastitis humana ha sido hasta la fecha una enfermedad subestimada, ya que su diagnóstico microbiológico no se realiza de forma sistemática, y habitualmente sólo se consideran mastitis los casos agudos que cursan con una sintomatología evidente. En este trabajo se revisa la literatura médica acerca de los posibles factores de riesgo que podrían estar implicados en el desarrollo de mastitis infecciosa, incluidos los aspectos relacionados con el historial médico de la madre y del hijo, el embarazo, el parto, el posparto y la lactancia. En este sentido, el hecho de profundizar en el conocimiento de dichos factores permitirá proporcionar un asesoramiento adecuado durante la lactancia, así como diseñar estrategias para prevenir esta enfermedad, con el objetivo final de que muchas parejas madre-hijo disfruten plenamente de los beneficios que proporciona la lactancia materna.
Deshidratación hipernatrémica asociada a la alimentación con lactancia materna en el periodo neonatal
Caries dental: influencia de los hábitos de higiene bucodental y de alimentación en niños en edad escolar
Consecuencias inesperadas de las iniciativas actuales de lactancia materna / Incidencia de las medicinas alternativas y complementarias en las tasas de vacunación de la gripe en niños estadounidenses
Los efectos adversos de las vacunas están claramente sobrepasados por sus beneficios. Pero, debido a la baja incidencia de estas enfermedades prevenibles gracias a las vacunas, la población puede tener una percepción errónea de que su riesgo es bajo, lo que provoca una participación relativamente baja en el programa de vacunación.
La vacuna de la gripe se recomienda para toda la población mayor de 6 meses, administrada anualmente. En los niños la vacuna es segura, y aunque su efectividad varía cada año, es inmunológicamente eficaz. Sin embargo, la vacunación de la gripe en niños estadounidenses es subóptima y sustancialmente más baja que la de otras vacunas.
El uso de las MAC en los niños no es raro. La estimación nacional de 2007 muestra que casi un 12% de los niños en Estados Unidos ha usado algún tipo de MAC en los últimos 12 meses. Este hecho es más común entre adolescentes, blancos no hispanos, con un alto nivel de educación de sus padres, una situación económica estable y un seguro de salud privado. La literatura disponible sugiere que es menos probable que se vacunen los niños que usan MAC, aunque no se ha estudiado cómo afecta esto a la vacuna de la gripe. El presente estudio examina la asociación de MAC con la vacuna de gripe en una muestra representativa de niños de Estados Unidos.
Se usaron los datos recogidos desde 2012 por el National Health Interview Survey (NHIS), acerca de la salud de la población no institucionalizada de Estados Unidos a través de entrevistas (contestadas por los padres) sobre unos 10.000 niños entre 4 y 17 años. Se estableció como variable dependiente si se habían puesto la vacuna de la gripe en el último año, y como variable independiente si habían utilizado alguna vez, por razones de salud, alguno de los 37 tipos de MAC en los últimos 12 meses. La prevalencia de haber usado alguna vez MAC variaba entre el 0,01 y el 6,4%, excepto el uso de multivitaminas/multiminerales (62,3%).
Los 37 tipos de MAC se agruparon en 4 categorías:
1. Sistemas de medicina alternativa (SMA; p. ej., acupuntura) (3,8%).
2. Terapias de base biológica (TBB; p. ej., suplementos dietéticos) (7,6%).
3. Terapias basadas en el cuerpo y manipulativas (TBCM; p. ej., manipulación quiropráctica) (7,3%).
4. Tratamiento mente-cuerpo (TMC; p. ej., yoga) (5,3%).
El 43% de los niños recibió la vacuna antigripal en los 12 meses previos. Se ponían menos vacunas los sujetos que habían utilizado alguna vez SMA o TBCM, y la tasa más alta de vacunados se observó entre los que habían usado multivitaminas/multiminerales. No había ninguna asociación significativa entre los que habían utilizado alguna vez TBB o TMC. Las tasas de vacunación eran menores en los casos siguientes: niños blancos no hispanos (menor por cada año de edad cumplido), los que no habían efectuado recientemente una revisión del niño sano, los que no tenían una enfermedad crónica grave, los que carecían de seguro médico y los que habían realizado menos visitas médicas recientemente.
Discusión
Aunque las MAC se usan mayoritariamente junto con la medicina convencional, el presente estudio provee una clara evidencia de que los niños de Estados Unidos que alguna vez han usado algún subtipo de TAM o TBCM tenían menores tasas de vacunación contra la gripe. Estos tipos de MAC requieren un contacto con los profesionales que las practican (a diferencia de los suplementos de hierbas, las dietas alternativas o el yoga), críticos respecto a las vacunas, en contra de ellas o que aconsejan calendarios vacunales distintos al recomendado por el gobierno federal.
Varias covariables estaban significativamente asociadas, entre ellas acudir con más frecuencia al pediatra, ser de menor edad o padecer asma. Generalmente, la educación parental más alta se asocia a una mayor tasa de vacunación gripal en niños, aunque en este estudio se encontró una asociación inversa.
La limitación fundamental de este estudio es que se excluyó a los menores de 4 años, el grupo de edad con mayor riesgo de desarrollar complicaciones asociadas a la gripe.
Lo que aporta este trabajo:
Es cada vez mayor el número de personas que usan terapias complementarias y alternativas, algunas veces frente a tratamientos o recomendaciones de la medicina convencional. En este estudio se pone de manifiesto que utilizar alguna de estas terapias (en especial las suministradas por otras personas) se asocia a tasas de vacunación de la gripe en niños inferiores a las del resto de la población. Hoy día la pregunta sobre las terapias alternativas y complementarias debería formar parte de la anamnesis en cualquier consulta médica y explicar que su empleo no debe contradecir datos evidentes como, por ejemplo, la eficacia de la vacunación de la gripe.
Análisis de minimización de costes y del impacto sobre el presupuesto del Sistema Nacional de Salud del tratamiento del TDAH con metilfenidato de liberación inmediata, en lugar de las diferentes alternativas de metilfenidato de liberación prolongada
Los alimentos orgánicos en la alimentación infantil
Comunicación científica (XXXVII). Cómo hacer una estrategia social media para pediatras (IV). Técnicas de «social customer relationship management» en redes sociales
Se explican las características de un customer relationship management (CRM) aplicado al mundo de la salud, en especial al de la pediatría, en un entorno online, lo que se denomina social CRM. Se exponen sus beneficios e inconvenientes, y se describen algunas tácticas que los pediatras están siguiendo para gestionar las comunidades de pacientes online, actuales y potenciales, que se generan en torno a sus cuentas profesionales en los medios sociales o redes sociales, explorando así la gestión online de la relación antes y después de la cita médico-paciente.
La web de un servicio médico como herramienta de información, formación y gestión
Galactocele: tumor mamario en un varón prepuberal
«Perineal groove», ese gran desconocido
El perineal groove, o surco perineal, es una malformación benigna y poco frecuente que tiende a la resolución espontánea. De forma general, se caracteriza por un surco húmedo en la línea media perineal que se extiende desde la horquilla vulvar hasta el ano. El surco perineal se presenta más frecuentemente en niñas, aunque también se ha descrito en varones. Se ignora su incidencia real, debido en parte al desconocimiento del mismo por parte de los profesionales. Presentamos el caso de una recién nacida de 2 días de vida en la que se detecta una lesión lineal en el periné. Es importante su reconocimiento dado que entraña un amplio diagnóstico diferencial de lesiones, incluidas algunas con importantes connotaciones legales por la zona en la que se presenta.
Enfermedades emergentes: eosinofilia en el niño migrante
Enterobacterias productoras de carbapenemasas en un hospital pediátrico
Deficiencia en vitamina D: un reto diagnóstico
Desde hace varios años la literatura científica prolifera en torno a un nuevo problema de salud que parece constatarse a escala mundial: el déficit de vitamina D. El establecimiento de nuevos y más bajos umbrales de deficiencia asumidos por la mayor parte de sociedades científicas internacionales, ante los insólitos vínculos que esta vitamina podría tener con la salud global de la población, ha generado multitud de estudios que publican cifras de prevalencia de déficit elevadas, y con una amplia variabilidad. La controversia sobre el nivel considerado como límite de suficiencia es especialmente evidente en la población infantil, en la que la investigación es más limitada. No obstante, una nueva hipótesis acerca de la fracción libre de la vitamina D como posible y óptimo marcador en la evaluación de su estatus está estimulando una novedosa e interesante línea de investigación. Tras mucha polémica bibliográfica parecía que ya teníamos una respuesta aceptable. La Academia Nacional de las Ciencias recogía en 2010, en su «Dietary references intakes for calcium and vitamin D», que «aunque el nivel sérico de 25-hidroxivitamina (25OHD) no está validado como sustituto de resultados en salud, puede servir como referencia», concluyendo que es razonable considerar 20 ng/mL como el umbral para casi toda la población1. «20» pasó a ser la cifra mágica. Pero la pregunta ahora ha cambiado: ¿es realmente la 25OHD total el parámetro que mejor mide la suficiencia en vitamina D?, ¿podría ser la fracción libre de 25OHD el marcador que cabría considerar? Encontrar una respuesta casi nunca es el fin de la búsqueda..., afortunadamente.
Comunicación científica (XXXVI). Cómo hacer una estrategia social media para pediatras (III). La «responsible research & innovation» (RRI) como análisis de la demanda potencial de pacientes y pares
La investigación e innovación responsable (responsible research & innovation [RRI]) busca que todos los actores sociales (investigadores, ciudadanos, responsables políticos, empresariado e industria, sector educativo) trabajen conjuntamente durante el proceso de investigación e innovación, con carácter anticipatorio y adaptativo a las necesidades de la sociedad. Con ese objetivo, hay diversas técnicas que ayudan al científico a obtener un impacto aplicado de su investigación lo antes posible en la mejora de la vida de los ciudadanos; ayudan a descubrir esas necesidades sociales, de forma que su investigación tenga, además, más probabilidades de suscitar interés entre sus pares, siendo así más citado. En el presente artículo se propone una técnica de identificación de temáticas de investigación y términos de búsqueda adaptados a la demanda de información, tanto de pacientes como de pares, basada en indicadores cibermétricos, con el fin de determinar líneas emergentes de investigación, priorizar contenidos científicos y establecer lazos de unión, tanto con la sociedad como con otros científicos.
Gran reconocimiento internacional a la formación en pediatría de Atención Primaria española

- Dos iniciativas españolas de formación en pediatría destacan en el Global Pediatric Educational Consortium (GPEC).
- El curriculum de Atención Pimaria (AP) basado en competencias ayuda a la formación del pediatra y del residente a nivel mundial.
- La adaptación al español del curriculum europeo facilitará la formación en AP a los pediatras en Sudamérica.
Madrid, 29 de agosto de 2016.- En el marco de la reunión del Global Pediatric Educational Consortium (GPEC) celebrada en Barcelona, se han dado cita, junto con el secretario de este grupo internacional, Hazel Ham, importantes representantes de la pediatría de nuestro país; entre ellos: la doctora Carmen Villaizán, vicepresidente de formación de la Asociación Española de Pediatría de Atención Primaria (AEPap) y representante de la Confederación Europea de Pediatras de Atención Primaria (ECPCP) y el doctor Ángel Carrasco, secretario de la Asociación Española de Pediatría (AEP).
El GPEC es una asociación de expertos internacionales en formación, capacitación y acreditación pediátricas, y tiene como principal objetivo conseguir la mejor formación para los residentes y para los pediatras a nivel mundial. Todos sus miembros comparten recursos y experiencia para aumentar así la cantidad y calidad de pediatras y subespecialistas, proporcionando los recursos formativos primordiales a todos aquellos países interesados, en especial a los que están en vías de desarrollo.
El ejecutivo de GPEC está formado por delegados que proceden de países como Estados Unidos, Egipto o Brasil entre otros. De los invitados a la reunión destacan Christiana Russ, responsable de formación del Children´s Hospital de Boston y de la Universidad de Harvard y Teresa Bandeira, presidenta de la Sociedad Portuguesa de Pediatría.
El objetivo principal de esta reunión fue profundizar en la implantación de un currículum pediátrico mundial, para formar a residentes de pediatría que sea flexible y basado en competencias.
En este sentido, la doctora Carmen Villaizán fue la encargada de presentar el currículum de formación en AP, realizado junto a la Confederación Europea de Pediatras de AP; y el doctor Ángel Carrasco, por su parte, presentó la plataforma de formación Continuun de la AEP. Ambos se adaptan al GPEC, y han tenido una buena acogida y han sido valorados muy positivamente por los asistentes.
Además, los representantes españoles fueron felicitados por la traducción del currículum GPEC al español, ya que esto ha permitido su aplicación en Sudamérica.
Dicha traducción fue realizada por la AEP, gracias al trabajo desinteresado del doctor Ángel Carrasco quien trabajó con 40 pediatras españoles, muchos de ellos pediatras de AP de la AEPap, la mayoría integrantes del grupo de formación MIR liderado por el doctor Miguel Ángel Fernández - Cuesta Valcarce y del grupo de pediatría basada en la evidencia.
Por su parte, el doctor Dioclecio Campos, vocal de Brasil de GPEC, se ha mostrado muy interesado en el currículum europeo, manifestando que le gustaría implantarlo en el Foro de Sociedades Pediátricas del Cono Sur (FCSPS), formado por Uruguay, Paraguay, Argentina, Chile, Bolivia y Brasil.
Desde la AEPap afirman estar muy orgullosos del alcance y reconocimiento mundial del trabajo de la doctora Carmen Villaizán y del grupo de docencia MIR con el currículum de formación y añaden que seguirán trabajando en contribuir a la mejora de la pediatría.
Comunicación científica (XXXV). Cómo hacer una estrategia social media para pediatras (II). Visibilidad en redes sociales
Los científicos, en general, han encontrado en las redes sociales una plataforma válida en la que apoyarse para aumentar la exposición de sus trabajos científicos a sus pares. Los pediatras, en particular, pueden usar las redes sociales con diferentes objetivos, según la red social en la que participen. Se estudian las razones por las que el pediatra debe participar en redes sociales, las diferencias entre ambos tipos de redes (generalistas frente a verticales o científicas) y cómo estimar la audiencia probable para conseguir optimizar los esfuerzos de visibilidad de los profesionales de pediatría en ambos tipos de redes o medios sociales, así como mejores prácticas en ambas categorías.
La variabilidad en el manejo de la bronquiolitis. ¿Por qué no hacemos lo que leemos?
Envenenamiento por picadura de alacrán. Presentación de un caso clínico
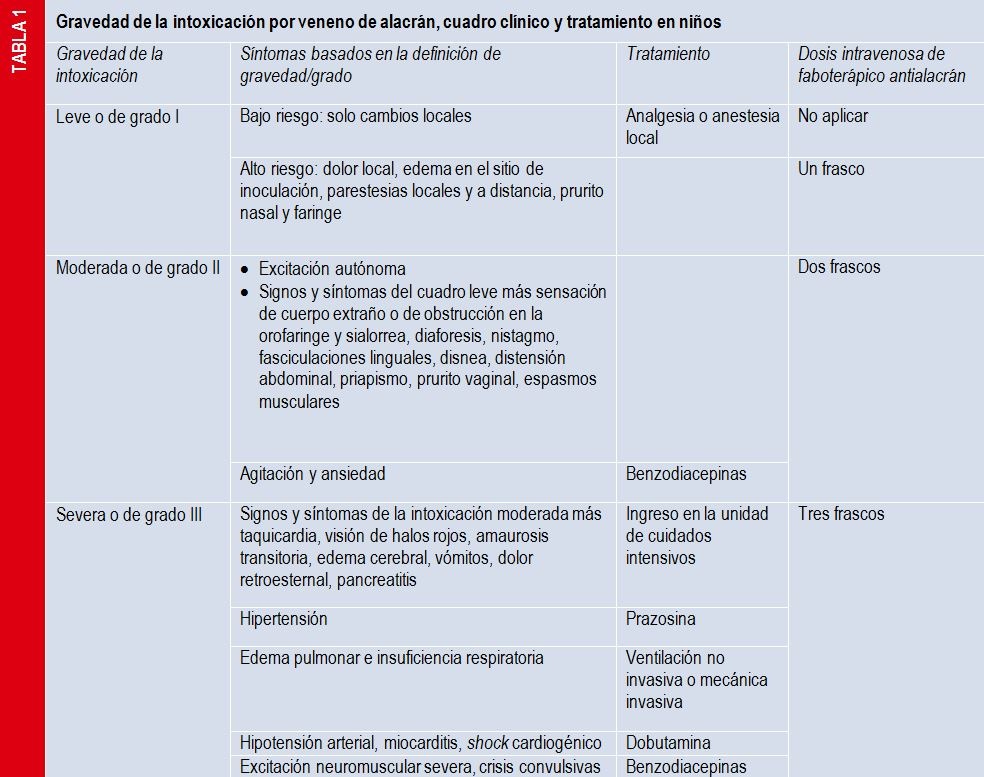
La cirugía plástica en adolescentes / Horas de sueño recomendadas en la población infantil
J Clin Sleep Med. 2016; 12: 785-786.
- Los lactantes entre 4 y 12 meses deberían dormir entre 12 y 16 horas al día de forma regular para tener una salud óptima. No hay recomendaciones en menores de 4 meses, debido a la amplia variabilidad en los patrones y en la duración del sueño a esa edad y su correlación con el estado de salud.
- Los niños entre 1 y 2 años deberían dormir entre 11 y 14 horas al día (incluyendo siestas) para tener una buena salud.
- Entre 3 y 5 años deberían dormir entre 9 y 12 horas (incluyendo siestas).
- Los niños de 6 a 12 años deberían dormir entre 9 y 12 horas diarias.
- Finalmente, los adolescentes entre 13 y 18 años deben dormir entre 8 y 10 horas de forma regular.
Unidad de Nutrición Clínica. Hospital Universitario «12 de Octubre». Madrid
El trastorno por déficit de atención con/sin hiperactividad en España: crónica de los últimos 15 años
El trastorno por déficit de atención con/sin hiperactividad tiene una gran relevancia en la infancia y puede perdurar en la adolescencia y en la edad adulta. Cada vez se conoce más y mejor en España esta afección, y desde hace más de una década su diagnóstico ha aumentado, así como los medios para su asistencia, tanto sanitaria como educativa. Tras hacer una breve historia de esta enfermedad, en esta revisión se actualizan los datos de prevalencia y del diagnóstico precoz, así como de los tratamientos disponibles actualmente. Asimismo, se comentan algunos aspectos de la realidad asistencial y cómo se está planificando el futuro.
Leishmaniasis cutánea y visceral en una lactante sana
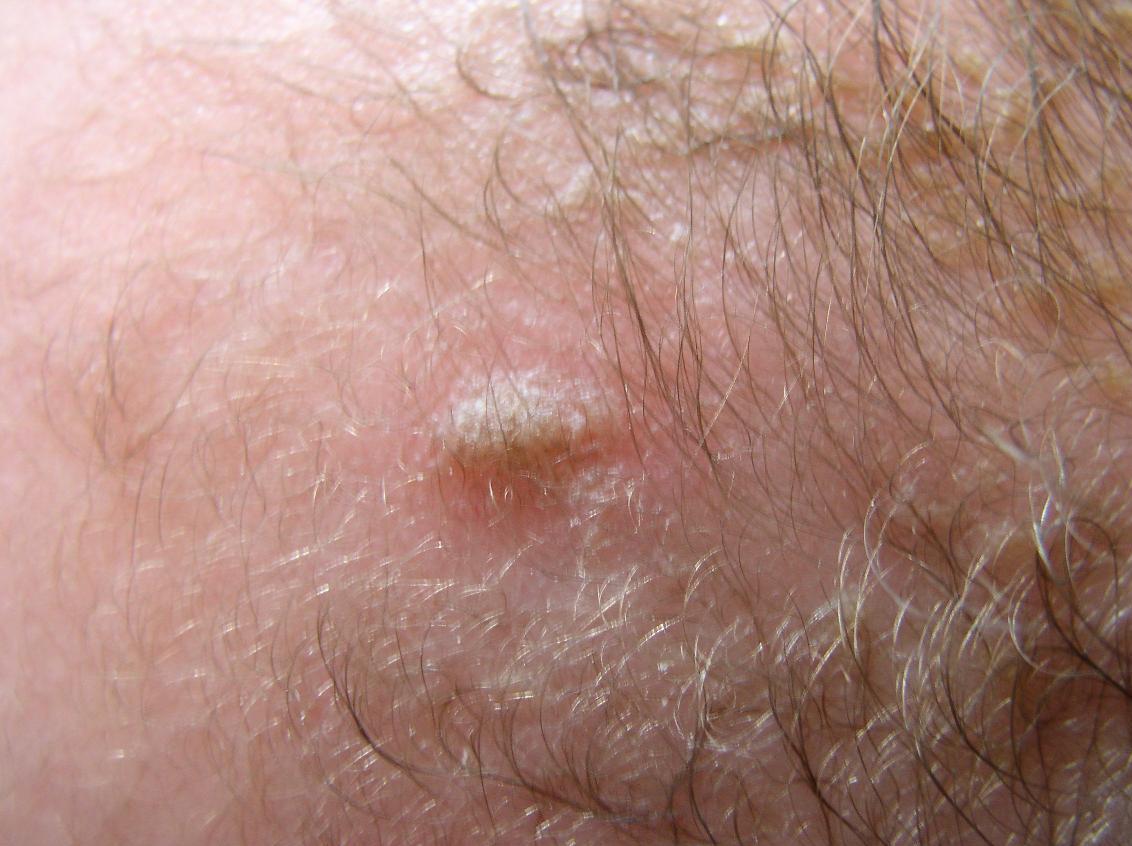 Presentamos el caso de una lactante sana, de 11 meses de edad, con una forma visceral de leishmaniasis, diagnosticada a raíz de una lesión cutánea que había pasado desapercibida. Acudió a nuestra consulta por la presencia de fiebre de 4 días de evolución, con decaimiento y rechazo parcial de la alimentación, sin otra sintomatología. En la exploración física presentaba palidez cutaneomucosa, hepatoesplenomegalia y una lesión papular eccematosa de 6-7 mm de diámetro en el cuero cabelludo, que los padres estaban tratando como dermatitis seborreica desde hacía 4-5 meses (figura 1). El hemograma mostró la presencia de leucopenia con neutropenia, anemia y trombopenia, sin observarse formas blásticas. La proteína C reactiva se encontraba elevada, con un valor de 186 mg/L, y el resto de la bioquímica era normal. Los resultados de la serología para el virus de Epstein-Barr, citomegalovirus y parvovirus B19 fueron negativos, y ante la sospecha clínica de leishmaniasis visceral se solicitó un frotis periférico para Leishmania. En sangre periférica no se observaban formas compatibles con el parásito, por lo que se decidió biopsiar la lesión inespecífica del cuero cabelludo y realizar un aspirado de médula ósea. Se inició tratamiento antibiótico de amplio espectro y con anfotericina B liposomal. La biopsia cutánea mostró abundantes formas de Leishmania en células mioepiteliales (figura 2), lo que confirmó el diagnóstico de leishmaniasis visceral y, probablemente, la lesión cutánea era el punto de inoculación. Se revisó la muestra de sangre periférica y se halló alguna forma aislada compatible con Leishmania; el aspirado medular no mostraba de forma clara la presencia de parásitos. Se completó el tratamiento con anfotericina B liposomal, 4 mg/kg/día en 5 dosis, y la sexta dosis administrada al décimo día. La evolución clínica y analítica de la paciente fue favorable. Durante el seguimiento de 18 meses la paciente se ha mantenido asintomática sin presentar ninguna recaída.
Presentamos el caso de una lactante sana, de 11 meses de edad, con una forma visceral de leishmaniasis, diagnosticada a raíz de una lesión cutánea que había pasado desapercibida. Acudió a nuestra consulta por la presencia de fiebre de 4 días de evolución, con decaimiento y rechazo parcial de la alimentación, sin otra sintomatología. En la exploración física presentaba palidez cutaneomucosa, hepatoesplenomegalia y una lesión papular eccematosa de 6-7 mm de diámetro en el cuero cabelludo, que los padres estaban tratando como dermatitis seborreica desde hacía 4-5 meses (figura 1). El hemograma mostró la presencia de leucopenia con neutropenia, anemia y trombopenia, sin observarse formas blásticas. La proteína C reactiva se encontraba elevada, con un valor de 186 mg/L, y el resto de la bioquímica era normal. Los resultados de la serología para el virus de Epstein-Barr, citomegalovirus y parvovirus B19 fueron negativos, y ante la sospecha clínica de leishmaniasis visceral se solicitó un frotis periférico para Leishmania. En sangre periférica no se observaban formas compatibles con el parásito, por lo que se decidió biopsiar la lesión inespecífica del cuero cabelludo y realizar un aspirado de médula ósea. Se inició tratamiento antibiótico de amplio espectro y con anfotericina B liposomal. La biopsia cutánea mostró abundantes formas de Leishmania en células mioepiteliales (figura 2), lo que confirmó el diagnóstico de leishmaniasis visceral y, probablemente, la lesión cutánea era el punto de inoculación. Se revisó la muestra de sangre periférica y se halló alguna forma aislada compatible con Leishmania; el aspirado medular no mostraba de forma clara la presencia de parásitos. Se completó el tratamiento con anfotericina B liposomal, 4 mg/kg/día en 5 dosis, y la sexta dosis administrada al décimo día. La evolución clínica y analítica de la paciente fue favorable. Durante el seguimiento de 18 meses la paciente se ha mantenido asintomática sin presentar ninguna recaída. Los mecanismos de inmunidad celular determinan la resistencia o susceptibilidad a la infección por Leishmania. La activación de las células T helper (Th) desempeña un papel importante en la respuesta inmunitaria frente al parásito, tal como se ha observado en el modelo murino de infección con L. major, L. braziliensis y L. infantum. Se han establecido 2 tipos de respuesta según se expresen las subpoblaciones Th1 o Th2. La resistencia a la infección está mediada por la expansión de los linfocitos Th1 que producen interferón gamma (IFN-γ) e interleucina 12 (IL-12); estos mediadores activan la destrucción de los macrófagos parasitados y, por tanto, de las Leishmania, lo que provoca la curación de la lesión. Por el contrario, la susceptibilidad a la infección se relaciona con la expansión de las células Th2 productoras de citocinas que inhiben la respuesta celular (IL-4, IL-5, IL-10)1,3. Los pacientes con leishmaniasis cutánea difusa o leishmaniasis visceral activa muestran una mínima o nula respuesta inmunitaria celular específica contra Leishmania, aunque estas respuestas se recuperan tras un tratamiento adecuado.
Los mecanismos de inmunidad celular determinan la resistencia o susceptibilidad a la infección por Leishmania. La activación de las células T helper (Th) desempeña un papel importante en la respuesta inmunitaria frente al parásito, tal como se ha observado en el modelo murino de infección con L. major, L. braziliensis y L. infantum. Se han establecido 2 tipos de respuesta según se expresen las subpoblaciones Th1 o Th2. La resistencia a la infección está mediada por la expansión de los linfocitos Th1 que producen interferón gamma (IFN-γ) e interleucina 12 (IL-12); estos mediadores activan la destrucción de los macrófagos parasitados y, por tanto, de las Leishmania, lo que provoca la curación de la lesión. Por el contrario, la susceptibilidad a la infección se relaciona con la expansión de las células Th2 productoras de citocinas que inhiben la respuesta celular (IL-4, IL-5, IL-10)1,3. Los pacientes con leishmaniasis cutánea difusa o leishmaniasis visceral activa muestran una mínima o nula respuesta inmunitaria celular específica contra Leishmania, aunque estas respuestas se recuperan tras un tratamiento adecuado.Un patrón de sueño alterado de los padres disminuye la calidad de sueño de los niños referido por los padres / Las experiencias sensoriales en etapas tempranas de la vida pueden influir en las preferencias y en la aceptación de los alimentos
Pediatrics. 2016; 137(4): e20153425.
La pediatría se caracteriza por el uso de información de segunda mano en muchas ocasiones, ya que son los padres los que refieren los problemas de sus hijos. Así ocurre, por ejemplo, con los problemas de sueño de los niños. Los autores de este estudio, de la Universidad de Turku en Finlandia, se plantean si un patrón de sueño alterado de los padres disminuye la calidad de sueño de los niños referido por los padres. La pregunta no es tanto si los niños duermen mal, sino si los padres que duermen mal piensan que sus hijos duermen mal.
Las madres depresivas cuentan más problemas de comportamiento de sus parientes que las madres no depresivas. Esta clase de distorsión puede llegar a ser problemática en condiciones médicas en las que el diagnóstico depende en gran medida de la descripción dada por los padres, como ocurre con las alteraciones del sueño en los más pequeños. Muchas de estas alteraciones se diagnostican basándose en lo que cuentan los padres; el uso de métodos objetivos es raro.
El objetivo de este estudio era analizar si la calidad de sueño de los padres se asociaba con la percepción de la calidad de sueño indicada por éstos en sus hijos, comparada con la medida objetiva de la calidad de sueño de los niños. Es decir, si los padres con una baja calidad de sueño comunican más dificultades relativas al sueño en sus hijos que lo sugerido por una valoración objetiva.
Para ello, los autores seleccionaron a 100 niños de 2-6 años de edad, que asistían a guarderías (la edad escolar en Finlandia comienza a los 7 años), mediante una carta de invitación a participar en el estudio, entre enero de 2014 y febrero de 2015. La actividad de los niños durante el sueño (medida de su calidad de sueño) se valoró por medio de un brazalete actígrafo (similar a un acelerómetro que graba los movimientos del niño, con lo que se estima el tiempo de sueño y los despertares) durante una semana. A los padres de los niños que aceptaron participar se les facilitó un diario de sueño y una escala de alteraciones del sueño de los niños de 30 preguntas (Sleep Disturbance Scale for Children). Los padres también cumplimentaron una escala de su propio sueño, de 4 preguntas (escala de Jenkins), junto con un cuestionario general de salud de 12 ítems (12-item General Health Questionnaire), así como preguntas sobre otros factores demográficos (nivel socioeconómico, bienestar familiar).
Los resultados mostraron que los padres que presentan más dificultades en el sueño indican más problemas de sueño en sus hijos. Esta asociación no pudo demostrarse con la medición objetiva de la calidad del sueño de los niños mediante el actígrafo. El cansancio de los padres actuó como un sesgo de observación con respecto al sueño de los niños.
Por tanto, una calidad peor del sueño de los padres se asoció a un sobrediagnóstico en los problemas de sueño de sus hijos. Este hallazgo pone de manifiesto que es importante considerar la calidad del sueño de los padres cuando se trata de diagnosticar, tratar o investigar las alteraciones del sueño de los niños.
Lo que este estudio aporta:
Los problemas de sueño en los niños son causa frecuente de consulta al pediatra y conllevan, con frecuencia, la realización de pruebas diagnósticas o de tratamientos en los niños con el fin de descartar enfermedades. Sin embargo, muchos de estos trastornos del sueño de los niños no se valoran con criterios objetivos, sino que se basan en la historia recogida de los padres. Este estudio pone de manifiesto que la comunicación de problemas del sueño en los niños es mayor en los progenitores que tienen problemas con su propio sueño, y no pueden refrendarse cuando se utilizan criterios objetivos.
The lasting influences of early food-related variety experience: a longitudinal study of vegetable acceptance from 5 month to 6 years in two populations
Maier-Nöth A, Schaal B, Leathwood P, Issanchou S.
PLoS One. 2016; 11(3): e0151356
Existe un interés creciente por conocer –y potenciar– la adquisición de habilidades relacionadas con la alimentación en el niño pequeño. Las experiencias sensoriales en etapas tempranas de la vida pueden influir en las preferencias y en la aceptación de los alimentos. Existen datos contrastados que muestran que tanto la lactancia natural como la exposición a una mayor variedad de verduras en el inicio de la alimentación complementaria se asocian a un mayor consumo de nuevos alimentos posteriormente.
En el estudio presentado se examinan los efectos a largo plazo en tres contextos de exposición a estímulos relacionados con las comidas: bebés amamantados, exposición temprana a una variedad de verduras y exposición repetida (en 8 comidas consecutivas) a verduras inicialmente rechazadas. Se evaluaron los resultados a los 15 meses, y a los 3 y 6 años de edad.
El estudio inicial se llevó a cabo en 147 parejas madre-hijo de dos localidades de Francia y Alemania. El inicio del seguimiento se realizó cuando los niños tenían 5,2 ± 0,1 meses, que es la edad habitual de comenzar con las verduras en dichas zonas. La oferta y aceptación de los alimentos a los 15 meses y a los 3 años se realizó por medio de cuestionarios que cumplimentaban los padres, mientras que el seguimiento a los 6 años se realizó en una sesión presencial en el laboratorio, con cuatro experimentos.
A los 15 meses, los participantes que habían recibido lactancia materna comían más verduras y les gustaba un mayor número de ellas que los que habían recibido una fórmula infantil. Las verduras que inicialmente rechazaron, pero que aceptaron después de una exposición repetida, continuaban formando parte de su dieta en un 79% de los niños. A los 3 años, continuaba gustándole al 73%.
En el experimento presencial realizado a los 6 años de edad se observó que, tanto los niños que habían sido amamantados como los que probaron una mayor variedad de verduras en el inicio de la alimentación complementaria, comían más verduras nuevas y les gustaban más. Tenían más interés también en probar verduras que los que habían recibido una fórmula o una escasa variedad de verduras. Y a los 6 años, un 57% de los niños que probaron verduras que inicialmente no les gustaban, continuaban consumiéndolas.
Los autores demostraban así que las influencias tempranas en la alimentación contribuyen a modificar los hábitos alimentarios y que ese efecto persiste a lo largo del tiempo.
Lo que este estudio aporta:
Este estudio confirma algo ya conocido: el tipo de lactancia y la experiencia con una mayor variedad de verduras en el momento de comenzar con la alimentación complementaria condiciona la aceptación posterior de otras verduras. Y, además, ese efecto persiste en el tiempo. Hay razones científicas que apoyan el consejo nutricional que se les da a las mamás respecto a cómo alimentar a sus hijos, y puede ayudarles a favorecer la adquisición de hábitos saludables en ellos.
J.M. Moreno-Villares
Unidad de Nutrición Clínica. Hospital Universitario «12 de Octubre». Madrid
Drogas recreativas actuales: ¿podemos hacer más para prevenir su consumo en los adolescentes?
En la prevención del consumo de drogas en los adolescentes, además de reclamar a nuestras autoridades medidas eficaces de control de la oferta, la clave está en una mayor implicación de los padres, los docentes y los profesionales sanitarios, tres pilares que contribuyen a reforzar la información y la resiliencia del adolescente y, por tanto, su capacidad para decidir. Se proponen unas pautas para mejorar desde estas perspectivas y se realiza una descripción de las drogas recreativas de actualidad.
La tecnología 2.0 al servicio de la salud materno-infantil
Ácido docosahexaenoico. ¿Un ácido graso omega-3 esencial?
El papel del ácido docosahexaenoico (DHA) ha sido ignorado durante muchos años. En los últimos 20 años se le ha prestado progresivamente mayor atención, hasta el punto de poderse considerar como esencial en el lactante y en determinadas patologías, al resultar poco eficiente su conversión a partir de sus precursores omega-3 (ácidos alfa-linolénico y eicosapentaenoico). Este artículo analiza la evolución en el tiempo de las ingestas de DHA recomendadas por los organismos internacionales en diversas edades, las cantidades de DHA en alimentos para lactantes fijadas por la legislación de la Unión Europea (UE), y las autorizaciones en la UE de declaraciones nutricionales y también sobre propiedades saludables en el cerebro y la visión.
Tolerancia y eficacia de una fórmula extensamente hidrolizada en lactantes con alergia a proteínas de leche de vaca mediada por IgE. Estudio JUNGLO (I): hidrolizado extenso de caseína
Evitar el legado de la enfermedad renal. Enfoque sobre la niñez
El Día Mundial del Riñón 2016 se centra en las enfermedades renales durante la infancia y la enfermedad renal del adulto que puede iniciarse en una edad temprana. La enfermedad renal crónica (ERC) en la infancia difiere de la de los adultos en que predominan las causas derivadas de anomalías congénitas y trastornos hereditarios; las glomerulopatías y la enfermedad renal asociada a diabetes mellitus son poco frecuentes. Además, muchos niños con una lesión renal aguda pueden desarrollar eventualmente secuelas que lleven a la hipertensión y la ERC durante la adolescencia o la vida adulta. Los niños nacidos prematuros o pequeños para su edad gestacional tienen un incremento del riesgo de desarrollar ERC durante su vida. Las personas con un alto riesgo al nacer o en la primera infancia deberán ser monitorizadas estrechamente para poder detectar a tiempo los signos tempranos de enfermedad renal, con el fin de proporcionar una prevención o un tratamiento efectivos. Es factible establecer una terapia eficaz en niños con ERC avanzada; existen evidencias que demuestran que los niños evolucionan mejor que los adultos cuando reciben terapia de reemplazo renal, incluyendo la diálisis y el trasplante, aunque sólo una minoría requiere este tratamiento. Debido a las inequidades en el acceso a la atención médica, es necesario hacer un esfuerzo para que los niños con una enfermedad renal, donde sea que vivan, puedan ser tratados de manera eficaz, independientemente de su ubicación geográfica y su situación económica. Nuestra esperanza es que el Día Mundial del Riñón pueda informar al público en general, a los gestores y a los profesionales de la salud sobre las necesidades y las posibilidades que existen en torno a la enfermedad renal en la infancia.
Aplicación del test electrofisiológico de ejercicio en pacientes con miotonía congénita
Unidad Pedagógica Hospitalaria: humanizar la enseñanza y la asistencia
En el concepto de «pedagogía hospitalaria» conviene tener presente cuatro ideas clave: a) la humanización actúa como epicentro de la atención sanitaria, especialmente desde el siglo xxi; b) cabe considerarla como una sección más del Servicio de Pediatría; c) está más allá de la medicina y más allá de la educación especial, y d) es mucho más que un aula hospitalaria, pues su labor se extiende más allá de sus paredes.
En este artículo comentamos algunos aspectos generales de las Unidades Pedagógicas Hospitalarias (UPH) de la Comunidad Valenciana y centramos nuestra atención sobre la labor efectuada por la UPH del Servicio de Pediatría del Hospital General Universitario de Alicante. Con nuestra experiencia fundamentamos una conclusión: toda UPH debe convertirse en instrumento docente y también terapéutico, totalmente integrado en el Servicio de Pediatría, y su labor es una buena oportunidad para iniciar el camino de «hospital líquido», entendiendo como tal el que sale de sus paredes y convive con la sociedad.
Comunicación científica (XXX). Cómo hacer un protocolo de investigación
Características de los niños con enfermedad tuberculosa en Meki (Etiopía): el reto del diagnóstico con recursos limitados
Implicación diagnóstica del nevus anémico en la neurofibromatosis



Comunicación científica (XXIX). De la eHealth a la mHealth. Apps en pediatría
La generalización del uso de dispositivos móviles ha propiciado el desarrollo de un enorme y variado catálogo de aplicaciones en continua regeneración que se ha extendido al ámbito de la salud y que auxilian en la práctica médica a los profesionales médicos, al tiempo que permiten el desarrollo de un papel proactivo por parte del paciente en las tareas de seguimiento. En este trabajo se describen el contexto y las características de estas aplicaciones en el campo de la salud, y se proporciona un listado con algunas de las más destacadas en el campo de la pediatría, útiles para la actualización de conocimientos, el auxilio en las labores de gestión y la consulta sobre diagnósticos, así como las más empleadas por los padres para facilitar el seguimiento y la monitorización. Asimismo, se ofrece un listado con los principales directorios y buscadores de aplicaciones para facilitar la búsqueda entre una oferta tan variada y cada día más amplia, además de las iniciativas nacionales e internacionales orientadas a valorar la calidad y la fiabilidad de estas aplicaciones.
El pediatra y las recomendaciones nutricionales en la mujer embarazada y que lacta
La alimentación y la actividad física antes y durante el embarazo afectan de forma importante a la salud de la madre y de su hijo. Además, el periodo de lactancia acarrea un aumento de las necesidades de energía y nutrientes para la madre. Existe cada vez un mayor número de datos científicos sobre la trascendencia de la alimentación en las primeras etapas de la vida y el riesgo de desarrollar posteriormente enfermedad, que se engloba en la noción de «la nutrición en los mil primeros días». La intervención nutricional preventiva debería comenzar en el periodo periconcepcional, prolongarse durante el embarazo y los primeros años de vida y continuar como un programa de desarrollo de hábitos de vida saludable. Durante estos periodos es preciso que la mujer consiga alcanzar una ingesta suficiente de determinados nutrientes, más que un aumento en el aporte energético total. El pediatra debe considerarse un agente de salud pública clave para mejorar los hábitos de salud de toda la población. Desde el Comité de Nutrición de la Asociación Española de Pediatría se ha considerado de interés que el pediatra conozca las recomendaciones actuales para la alimentación de la mujer embarazada y lactante, con el fin de ejercer su influencia sobre la salud del recién nacido y del lactante.
Recomendaciones para subsanar los efectos negativos del sedentarismo en el trabajo / Tratamientos para prevenir y eliminar los piojos
Lo que aporta este trabajo:
Las condiciones físicas del trabajo (lugar, tiempo sentado, actividad física) pueden influir en la salud, pero también en el bienestar de los trabajadores. En una etapa en que las nuevas tecnologías han hecho disminuir mucho la actividad física en el trabajo, es preciso desarrollar estrategias novedosas para corregir esa desviación.
Esofagitis herpética en un adolescente inmunocompetente. A propósito de un caso
La esofagitis herpética se ha documentado ampliamente en pacientes inmunodeficientes, pero en pocas ocasiones se ha descrito en pacientes pediátricos inmunocompetentes. La gran mayoría de infecciones se deben al virus herpes simple tipo 1, aunque también se han descrito casos causados por el virus herpes simple tipo 2. Su aparición puede deberse a una reactivación del virus o, con menor frecuencia, a una primoinfección herpética. Las manifestaciones clínicas características son fiebre, odinofagia y dolor retroesternal. En pacientes inmunocompetentes, suele manifestarse como una enfermedad aguda autolimitada. La realización de una endoscopia alta permite establecer el diagnóstico de sospecha de la esofagitis herpética. Las imágenes macroscópicas suelen mostrar eritema y lesiones ulcerosas en la mucosa esofágica, y el estudio anatomopatológico cuerpos de inclusión intranucleares Cowdry tipo A. El diagnóstico se confirma mediante cultivo o reacción en cadena de la polimerasa para el virus en la muestra de biopsia. El tratamiento con aciclovir en pacientes sanos es controvertido, ya que es un proceso autolimitado. Presentamos un caso de esofagitis herpética de un adolescente inmunocompetente que se inició de forma aguda con síntomas severos del tracto digestivo alto. Es importante tener en cuenta esta entidad ante la presentación de casos agudos de odinofagia.
Comunicación científica (XXVIII). Nuevas formas de difusión de contenidos: streaming, webcasting y podcasting
Comunicación científica (XXVII). La propiedad intelectual, los permisos de reproducción, citación o transformación del contenido y los derechos de imagen
Dada la doble vertiente de los profesionales de la salud como consumidores y productores de contenidos científicos, es recomendable conocer los derechos de autor de las obras, así como los diferentes permisos para el uso de las mismas. Este artículo tiene como objetivo presentar algunas consideraciones que cabe tener en cuenta sobre la propiedad intelectual y la posibilidad de ofrecer los contenidos bajo una licencia Creative Commons, que permita que el autor ceda algunos derechos sobre la obra al usuario.
Lactancia materna y deshidratación neonatal. ¿Se puede disminuir el número de casos?
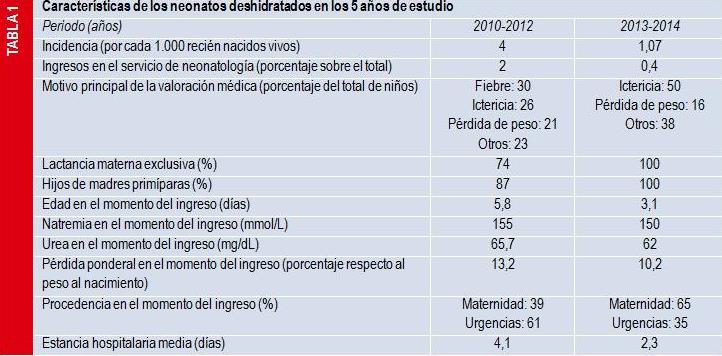
1. Ip S, Chung M, Raman G, Chew P, Magula N, DeVine D, et al. Breastfeeding and maternal and infant health outcomes in developed countries. Evid Rep Technol Assess (Full Rep). 2007; 153: 1-186.
2. Agostoni C, Braegger C, Decsi T, Kolacek S, Koletzko B, et al; ESPGHAN Committee on Nutrition. Breast-feeding: a commentary by the ESPGHAN Committee on Nutrition. J Pediatr Gastroenterol Nutr. 2009; 49: 112-125.
3. Melissa Bartick, Arnold Reinhold. The Burden of suboptimal breastfeeding in the United States: a pediatric cost analysis. Pediatrics. 2010; 125: e1048-e1046. Disponible en: http://pediatrics.aappublications.org/content/125/5/e1048.full.pdf
4. Unal S, Arhan E, Kara N, Uncu N, Aliefendioğlu D. Breast-feeding-associated hypernatremia: retrospective analysis of 169 term newborns. Pediatr Int. 2008; 50(1): 29-34.
5. Peñalver Giner O, Gisbert Mestre J, Casero Soriano J, Bernal Ferrer A, Oltra Benavent M, Tomás Vila M. Deshidratación hipernatrémica asociada a lactancia materna. An Pediatr (Barc). 2004; 61(4): 340-343.
6. Boskabadi H, Maamouri G, Ebrahimi M, Ghayour-Mobarhan M, Esmaeily H, Sahebkar A, et al. Neonatal hypernatremia and dehydration in infants receiving inadequate breastfeeding. Asia Pac J Clin Nutr. 2010; 19(3): 301-307.
7. Pelleboer RA, Bontemps ST, Verkerk PH, Van Dommelen P, Pereira RR, Van Wouwe JP. A nationwide study on hospital admissions due to dehydration in exclusively breastfed infants in the Netherlands: its incidence, clinical characteristics, treatment and outcome. Acta Paediatr. 2009; 98(5): 807-811.
8. Aguayo Maldonado J, Arana Cañada Argüelles C, Arena Ansótegui J, Canduela Martínez V, Flores Antón M, Gómez Papí A, et al. IHAN Calidad en la asistencia profesional al nacimiento y la lactancia. Informes, estudios e investigación. Madrid: Ministerio de Sanidad, Política e Igualdad. Centro de publicaciones, 2011.








