
Sr. Director:
La mancha mongólica (MM), también llamada melanocitosis dérmica congénita, suele aparecer en el nacimiento o durante las primeras semanas de vida. Aumenta en los 2 primeros años y después desaparece de modo gradual. A los 10 años la mayoría de estas manchas han remitido; si la mancha se mantiene en la edad adulta, se denomina MM persistente (3-4% de los orientales). Su frecuencia, similar en ambos sexos, varía entre los distintos grupos raciales. El término «mancha mongólica» se debe a su frecuencia elevada en las razas orientales, sobre todo en los mongoles, en quienes aparece en el 90% de los recién nacidos. Se observa en el 80% de los individuos de raza negra, en el 40% de los latinos y en menos del 10% de los caucasianos y judíos1-3.
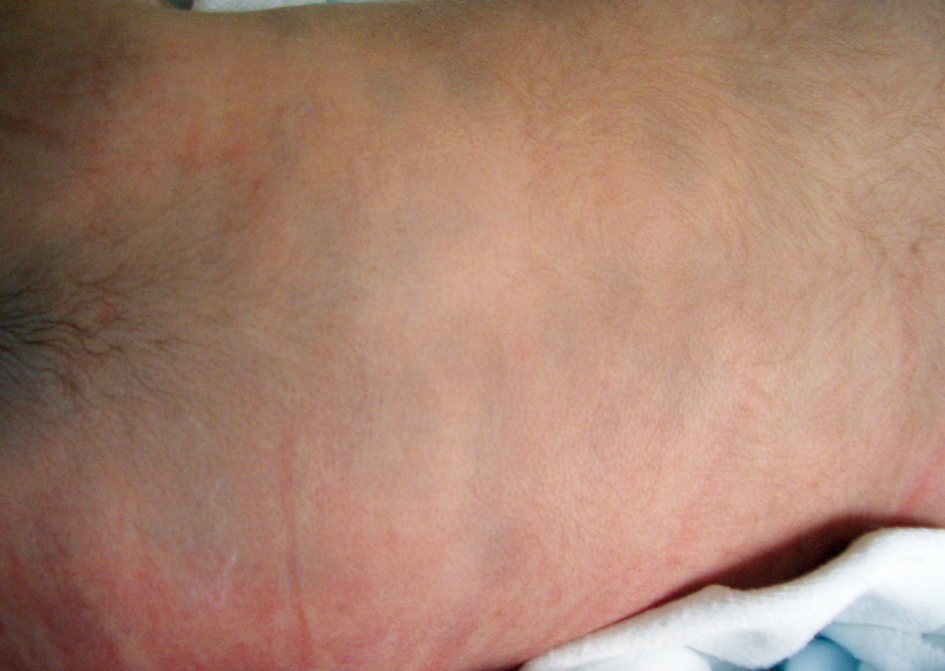
La localización clásica es la región lumbosacra y las nalgas. Se conoce como MM aberrante cuando se presenta en áreas atípicas, como la espalda, los hombros, el cuero cabelludo y las extremidades (figuras 1 y 2). Ésta es más probable que persista en la edad adulta. La forma extensa o generalizada no es excepcional, ya que aparece en más del 3% de los niños asiáticos, indios americanos y negros3-5.
Clínicamente, se presenta como una o varias máculas de morfología angulada, redonda u ovalada. El tamaño varía entre 1 y 20 cm y los bordes están mal definidos (las más grandes están mejor delimitadas). Tienen una coloración homogénea azul grisácea (figura 1) que no se acentúa en la exploración con lámpara de Wood. En personas de piel oscura adopta un tono verdoso (figura 2). La MM sobreimpuesta es una variante con zonas más oscuras en su seno3,6.
El estudio histológico muestra melanocitos dendríticos fusiformes dispersos entre los haces de colágeno de los tercios inferiores de la dermis. Esta melanocitosis dérmica se debe a que no se ha completado el proceso de migración de los melanocitos desde la cresta neural a la unión dermoepidérmica. Normalmente no hay melanocitos dérmicos desde la semana 20 de gestación. Las diferencias raciales podrían indicar la existencia de factores genéticos implicados en este proceso. La coloración azulada de las lesiones se debe al fenómeno de Tyndall: la pigmentación dérmica se ve azul, por la menor reflectancia de la región con longitudes de onda mayores en comparación con la zona que la rodea2,3.
No se han descrito casos de malignización de las MM. Dado que las máculas desaparecen con el paso de los años, no suelen necesitar tratamiento. En las lesiones persistentes se puede emplear maquillaje o laserterapia (con una mejor respuesta en MM aberrantes de pacientes de menos de 20 años)7.
La mayoría de las MM no tienen significación patológica. En ocasiones la presencia de formas aberrantes, extensas o múltiples puede facilitar el diagnóstico y posterior tratamiento precoz de ciertas enfermedades8:
1. Facomatosis pigmentovascular. Consiste en la combinación de una malformación vascular (casi siempre una mancha en vino de Oporto) con un lesión hiperpigmentada cutánea congénita (nevo epidérmico, nevo spilus o melanocitosis dérmica). Puede existir un nevo anémico. Se han clasificado en cinco tipos, que a su vez se han subdividido en a o b, en función de la ausencia o presencia de manifestaciones extracutáneas (como los síndromes de Sturge-Weber y de Klippel-Trenaunay). Aparece una MM en: a) facomatosis pigmentovascular tipo II (la más frecuente) asociada a una malformación capilar tipo mancha en vino de Oporto; b) tipo IV combinada con una mancha en vino de Oporto y un nevo spilus, y c) tipo V (facomatosis cesiomarmorata), que coexiste con cutis marmorata telangiectásica congénita4.
2. Gangliosidosis GM1, tipo 1 o infantil. Enfermedad debida a una deficiencia de la betagalactosidasa. En los primeros 6 meses de vida los pacientes presentan facies tosca, cuello corto, hipertrofia gingival, macroglosia, hipotonía, hipertricosis, retraso psicomotor y del desarrollo, convulsiones, hepatosplenomegalia, deformación de los cuerpos vertebrales, de la silla turca, anomalías de los huesos largos y de la pelvis, y manchas rojo cereza en la mácula. Los niños raramente sobreviven más de 3 años5,8.
3. Síndrome de Hurler. Mucopolisacaridosis tipo I, la más frecuente, producida por un déficit de alfa-L-iduronidasa. Comparte muchos rasgos con la gangliosidosis GM1 infantil. Alrededor de los 6 meses se observa opacidad corneal, visceromegalia, disostosis múltiple y rasgos faciales anormales. Provoca retraso mental, infecciones respiratorias y cardiopatía mortal alrededor de los 10 años de edad8.
4. Síndrome de Hunter. Mucopolisacaridosis tipo II, causada por un déficit de la enzima iduronato-2-sulfatasa y con un espectro de gravedad amplio. La forma grave cursa con síntomas similares al síndrome de Hurler9.
5. Labio leporino. Muchos niños japoneses con este defecto presentan una MM en la hendidura.
Se han descrito casos aislados en pacientes con enfermedad de Niemann-Pick, alfamanosidosis, síndrome de Sjögren-Larsson, hemangioma congénito, manchas café con leche (facomatosis pigmentopigmentaria), dedos supernumeraciones y melanosis leptomeníngea. Quizás algunas de estas asociaciones puedan ser casuales4,8.
El diagnóstico de la MM es fácil de establecer cuando la morfología clínica y la localización son típicas. En otras localizaciones podría confundirse con las siguientes entidades:
• Otras melanocitosis dérmicas, como el nevo azul (rara vez congénito y de localización muy variada), el nevo de Ota (sobre todo en la región periorbitaria, la sien, el área malar y la esclerótica ipsolateral), el nevo de Ito (regiones supraclavicular, escapular o deltoidea) y el hamartoma melanocítico dérmico (nalgas, manos y piernas)3.
• Otras neoplasias melanocíticas benignas, como la mancha café con leche, la melanosis de Becker y el nevo melanocítico congénito. El nevo melanocítico gigante, cuando afecta a la zona baja de la espalda y las nalgas, adopta un aspecto «en traje de baño». Difieren de la MM en el color y en los bordes bien definidos.
• Malformaciones vasculares, como las malformaciones glomovenosas o las manchas en vino de Oporto (se asocian a la MM en la facomatosis pigmentovascular) o los «precursores» de hemangiomas.
• Contusiones o hematomas. Éstos, si aparecen en lactantes de menos de 9 meses de edad y se sitúan en regiones típicamente protegidas, como las nalgas o la espalda, representan los signos físicos de maltrato infantil más frecuentes. Por tanto, una MM no diagnosticada o el desconocimiento de la variante aberrante, múltiple o sobreimpuesta podrían generar una sospecha errónea de maltrato. Cabe resaltar que el 40% de los neonatos de etnia gitana presentan MM (población con mayor incidencia de sufrir malos tratos según algunos estudios). Es curioso que el 12,5% de las madres atribuyan dichas manchas a un traumatismo durante el parto1. Por ello, es necesario anotar en la historia neonatal la presencia y la localización de las MM y proporcionar una correcta información a los padres10.
• Otras: argiria, ocronosis, eritema discrómico persistente (dermatosis cenicienta), hiperpigmentación postinflamatoria, hipermelanosis nevoide lineal y espiral, atrofodermia idiopática de Pasini y Pierini, y erupción fija medicamentosa.
En conclusión, la población española actual cuenta con más grupos raciales en que es frecuente la aparición de MM. Es importante conocer sus variantes (aberrante, persistente, múltiple, extensa o generalizada y sobreimpuesta), las enfermedades asociadas (facomatosis pigmentovascular, labio leporino y metabolopatías) y evitar su confusión con las lesiones de maltrato infantil.
Bibliografía
- Ikizoglu T, Ergör S, Asar GM, Yilmaz O. Frequency and characteristics of Mongolian spots among Turkish children in Aegean region. Turk J Pediatr. 2006; 48: 232-236.
- Avilés Izquierdo JA, Hernanz Hermosa JM, De la Cueva Dobao P. Mancha mongólica. Acta Pediatr Esp. 2004; 62: 60-61.
- Valdés F, Ginarte M, Toribio J. Melanocitosis dérmicas. Actas Dermosifiliogr. 2001; 92: 379-388.
- Torrelo A, Zambrano A, Happle R. Large aberrant Mongolian spots coexisting with cutis marmorata telangiectatica congenita (phacomatosis pigmentovascularis type V or phacomatosis cesiomarmorata). J Eur Acad Dermatol Venereol. 2006; 20: 308-310.
- Torrelo A, Solana LG, Mediero IG, Ruiz-Falco ML, García-Peñas JJ, Zambrano A. Mancha mongólica generalizada en un niño con gangliosidosis GM1 tipo 1. Actas Dermosifiliogr. 2000; 91: 31-33.
- Egemen A, Leung AK, Robson WL. Superimposed Mongolian spots. Pediatr Dermatol. 2008; 25: 233-235.
- Kagami S, Asahina A, Watanabe R, Mimura Y, Shirai A, Hattori N, et al. Laser treatment of 26 Japanese patients with Mongolian spots. Dermatol Surg. 2008; 34: 1.689-1.694.
- Ashrafi MR, Shabanian R, Mohammadi M, Kavusi S. Extensive Mongolian spots: a clinical sign merits special attention. Pediatr Neurol. 2006; 34: 143-145.
- Ochiai T, Suzuki Y, Kato T, Shichino H, Chin M, Mugishima H, et al. Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): a survey among 52 Japanese patients. J Eur Acad Dermatol Venereol. 2007; 21: 1.082-1.085.
- Al Jasser M, Al-Khenaizan S. Cutaneous mimickers of child abuse: a primer for pediatricians. Eur J Pediatr. 2008; 167: 1.221-1.230.








